Molecular epidemiology
In total, 1108 patients with skeletal abnormality from the DISCO study underwent ES. We identified eight probands with dual molecular diagnosis, including three (0.7%) from individuals with EOS, three (0.5%) from individuals with short stature, and two (2%) from individuals with CHFA (Table 1). All the eight probands have dual diagnosis of two autosomal dominant (AD) diseases. A total of 16 variants in 12 genes were identified. A substantial rate (5 of 10) of the identified causal variants were of de novo origin. The frequently observed molecular diagnoses (observed in more than one patient) include Osteogenesis Imperfecta Type I (COL1A1, MIM:166200), Neurofibromatosis, Type I (NF1, MIM:162200) and Marfan Syndrome (FBN1, MIM:154700).
The complex clinical features of patients with dual molecular diagnosis
Patients with dual molecular diagnosis often present blended phenotypes of two diseases, which significantly complicate their diagnostic odysseys. Here we report the detailed clinical characteristics of these patients to demonstrate the effect dual molecular diagnosis on the diagnosis of skeletal abnormality.
Case 1
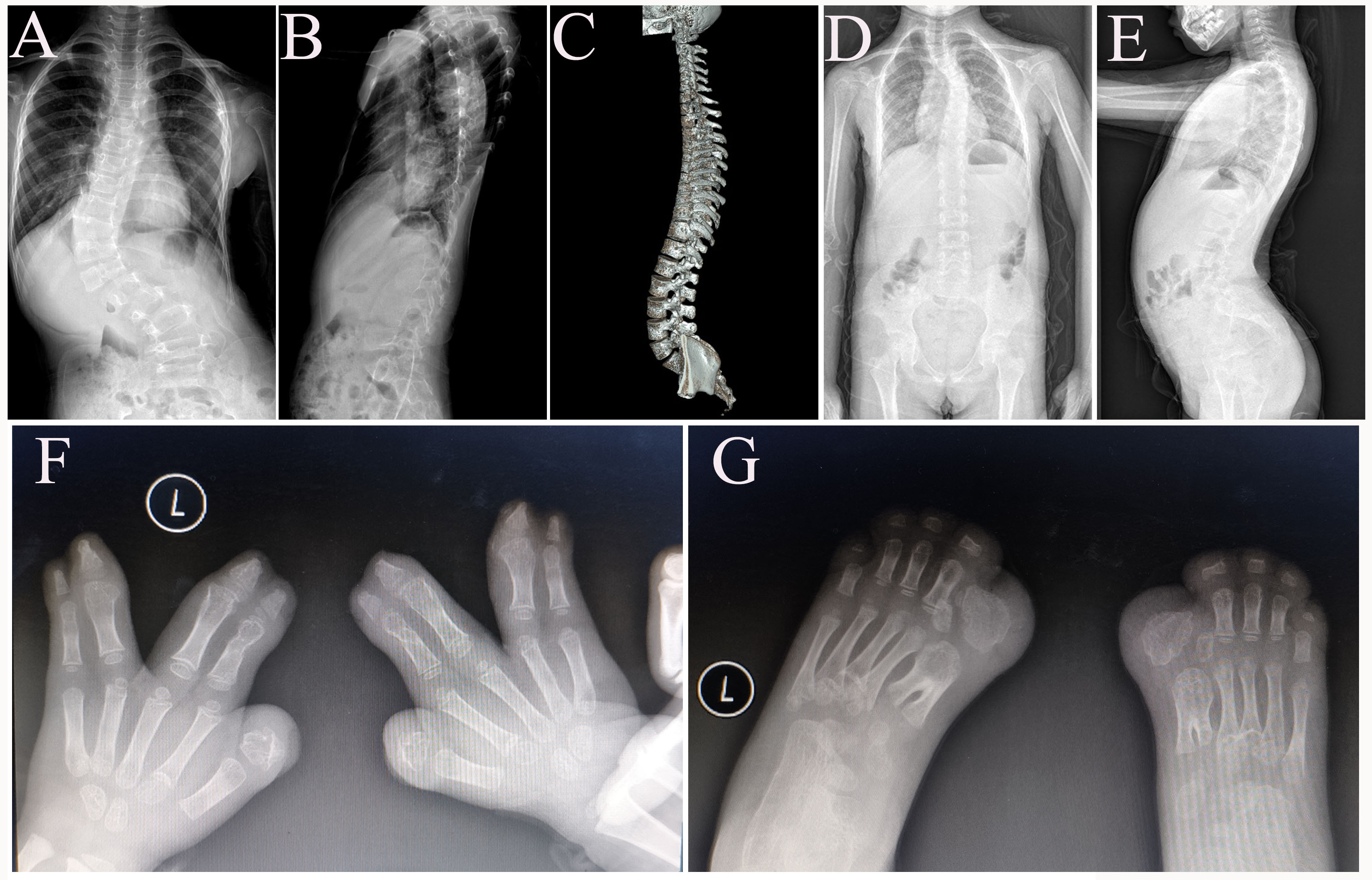
Patient DISCO-SCO2003P1972 was a 7-year-old boy with early-onset scoliosis (Figure 1.A, Supplementary Figure 1.A-B). At 2 years old, he was diagnosed with congenital dislocation of hip joint (Table 1) and underwent a surgical reduction. Meantime, a slight scoliosis was found during the hospitalization. At 7 years old, congenital scoliosis with segmentation failure of T10-L1 was identified through CT examination (Figure 1.A, Table 1). Physical examination showed disproportionate ocular hypertelorism and intellectual disability (Figure 1.A, Table 1). ES revealed a pathogenic heterozygous nonsense variant (c.2649G>A p.Trp883Ter) in FBN1 (Table 1), which is associated with Marfan syndrome (MIM:154700) (3). Consistently, the proband also presented Marfan syndrome-related phenotypes such as mitral valve prolapse, mild arachnodactyly and scoliosis. This variant was inherited from his mother, who had severe scoliosis, arachnodactyly and long slender limbs (Figure 1.A). Through further analysis of the exome data, a de novo variant in POGZ (c.1180_1181del p.Met394ValfsTer9) (Table 1) was found in the proband. This variant was previously reported and was associated with White-Sutton syndrome (WHSUS) (MIM:616364) characterized by intellectual disability, ocular abnormalities and skeletal deformities (12), which largely overlapped with the phenotypes of this patient. Therefore, we suggested that the complex phenotypes of this patient could be explained by a combined effect of variants in POGZ and FBN1.
Case 2
In case 2, the proband (DISCO-SCO1908P0067) was first referred to the clinic due to scoliosis at the age of fifteen years. The patient was clinically diagnosed as adolescent idiopathic scoliosis (AIS) (Supplementary Figure 1.C), i.e., scoliosis with unknown etiology (Table 1). He also presented pectus carinatum (Table 1). He experienced fracture twice at the age of 10 and 13. ES revealed that he carried two pathogenic variants, including a de novo variant in COL1A1 (c.1081C>T p.Arg361Ter) and a maternally inherited variant in FBN1 (c.1453C>T p.Arg485Cys) (Table 1). These two reported variants led to dual molecular diagnosis consisting of osteogenesis imperfecta type I and Marfan syndrome(13, 14). Although reported patients with osteogenesis imperfecta type I often present short stature (15), the stature of this patient is relatively high tall (Height: 185cm) (Figure 1.B , Table 1), which might be related with his second diagnosis of Marfan syndrome. Although FBN1 variant has been reported and considered as a pathogenic variant (14), no other features of Marfan Syndrome such as dolichostenomelia, arachnodactyly, joint laxity, velvety skin, ectopia lentis and cardiovascular manifestations were identified. This patient exemplified the apparently contrary effects of two monogenic disease on the same trait (height in this case).
Case 3
Patient DISCO-PCT2007P0019 was an 8-year-old girl. The proband suffered from right tibia fracture at the age of 2 years. At around the age of 6 years, the patient developed razorback deformity and unbalanced shoulder, accompanied with mild scoliosis. A progressive scoliosis was observed in the next 2 years (Supplementary Figure 1.D-E). A series of blended clinical phenotypes, such as blue sclera, cafe-au-Lait macules were observed as well (Figure 1.C, Table 1). We found vertebral malformations (T6, T7) in this patient (Figure 1.C) through radiographic examinations. Her father and younger brother presented with blue sclera but without any history of bone fracture. ES identified the one pathogenic variant in NF1 (c.2307del p.Thr770LeufsTer21) and another pathogenic variant in COL1A1 (c.2028+4A>G) (Table 1), both transmitted from her father. The NF1 variant could lead to a frameshift of NF1 (Table 1) and thus a loss-of-function effect, which is associated with neurofibromatosis type I and could explain the cafe-au-Lait macules in this patient (Figure 1.C, Table 1). The COL1A1 splicing variant (c.2028+4A>G) has been previously described to cause osteogenesis imperfecta (16), which could explain the recurrent bone fracture history in this patient. Interestingly, both neurofibromatosis type I and osteogenesis imperfecta are associated with scoliosis but with incomplete penetrance (15, 17). Therefore, the scoliotic phenotype in this patient might be caused by the synergistic effects of the dual molecular diagnosis.
Case 4
In this case, we reported a 6-year-old boy (DISCO-WAX677) with short stature and craniofacial abnormalities including depressed nasal bridge and long philtrum (Table 1). Then the patient was detected to be double heterozygote for putatively pathogenic ANKRD11 (c.2508dup p.Leu837ThrfsTer81) and COL11A1 (c.1180_1181del p.Met394ValfsTer9) variants on WES (Table 1). The ANKRD11 and COL11A1 variants were confirmed as de novo and paternal status, separately (Table 1). The clinical findings of the present case are compared with the reported phenotypes of KBG syndrome (MIM:148050) and Marshall syndrome (MIM:154780), caused by ANKRD11 and COL11A1 variants, respectively. A comparison between the findings in the patient and reported phenotypes of KBG and Marshall syndrome indicated overlapping clinical features in this case.
Case 5
This patient (DISCO-AX168) was an 8-year-old girl with short stature and developmental delay. She also presented widespread cafe-au-Lait macules and lumbar scoliosis (Figure 1.D, Table 1). Additionally, her father (162cm) and mother (140 cm) were both presented with short stature. Brain magnetic resonance (MR) was performed and abnormality of the cerebral white matter was identified. According to previously study (18), combined with MR results and family history, the patient was diagnosed with idiopathic short stature (ISS). Subsequently, the girl was sequenced and two pathogenic variants (Table 1), including a splicing variant in the NF1 gene (c.6705-1G>A) and a frameshift variant in the GLI2 gene (c.1189del p.Val397CysfsTer124) were identified. Variants in GLI2 have been shown to cause short stature, abnormal development of brain structures, hypopituitarism and facial dysmorphism in Culler-Jones syndrome (MIM:615849)(19). We suggested this patient’s presentation represents a mixture of distinct phenotypes, i.e., Cafe-au-Lait spots for NF type 1 (NF1, MIM:162200) and short stature for Culler-Jones syndrome (MIM:615849).
Case 6
Case 6 (DISCO-AX282) was a 12-year-old girl with short stature (Table 1). She also presented mild scoliosis, low posterior hairline, hyper pigmentation and webbed neck (Figure 1.E, Table 1). ES was performed to find the potential molecular etiology. A pathogenic nonsense variant in TP63 (c.109C>T p.Arg37Ter) and another pathogenic missense variant in PTPN11 (c.1510A>G p.Met504Val) were identified (Table 1). On the basis of published studies (20, 21), as well as phenotypes in this patient, we concluded this patient carried variants in both TP63 and PTPN11, resulting in a blended phenotype of Rapp-Hodgkin syndrome (MIM: 129400) and Noonan syndrome (MIM: 163950). Overlapping phenotypes (especially craniofacial malformations) of these two syndromes make the diagnosis challenging if solely based on clinical features.
Case 7
A 3-year-old proband (DISCO-RDD2001P0005) presented at birth with widespread interphalangeal joint contractures of hand (Figure 1.F) and atrial septal defect (Table 1). His father also experienced hand contracture deformities during his childhood. ES revealed a heterozygous variant (c.3437A>G p.Tyr1146Cys) in FBN2 gene and a heterozygous ANKRD11 variant (c.3024_3025del p.Lys1009GlyfsTer8), both with unknown origin (Table 1). The FBN2 variant could lead to Beals syndrome (Congenital contractual arachnodactyly) (MIM:121050), which is characterized by hand interphalangeal joint contractures (22, 23); The ANKRD11 mutation could cause KBG syndrome, which could explain the atrial septal defect in this patient (24). Therefore, this patient present mixed phenotypes of two distinct Mendelian disorders.
Case 8
This case is a 5-year-old male (DISCO-P19009402) with complex phenotypes. He presented congenital syndactyly of hand and foot (Supplementary Figure 1.F-G), cloverleaf skull, orbital hypertelorism, proptosis and midfacial hypoplasia (Table 1). He also had a history of hyperthermia during general anesthesia. ES identified a de novo missense variant (c.755C>G p.Ser252Trp) in FGFR2 gene and a paternally inherited frameshift variant in RYR1 gene (c.12788_12793dup p.Glu4263_Gly4264dup) (Table 1). The FGFR2 variant occurred in a known Apert syndrome hotspot (25). As reported, Apert syndrome was characterized by craniosynostosis, proptosis, midfacial hypoplasia and severe syndactyly of the hands and feet (25), which are concordant with the phenotypes of this patient. Pathogenic RYR1 variants are associated with malignant hyperthermia susceptibility 1(MHS 1) (MIM:145600) (26), which could explain the hyperthermia history in the patient.
Table 1
Summary of the clinical and molecular findings of studied subjects
|
Case Number
|
Case ID
|
Age
|
Sex
|
Inheritance
|
Gene
|
Clinical diagnosis
|
Molecular diagnosis
|
Zygosity
|
Transcript numbers
|
Origin
|
Variant type
|
Variant
|
GnomAD frequency
|
Gerp++
|
CADD
|
Patient phenotype
|
|
Case 1
|
SCO2003P1972
|
7
|
M
|
AD
|
POGZ
|
CS II
|
White-Sutton Syndrome
|
Het
|
NM_015100.3
|
de novo
|
Frameshift
|
c.1180_1181del p.(Met394ValfsTer9)
|
0
|
3.36
|
47.4
|
ocular hypertelorism; intellectual disability; scoliosis;congenital dislocation of hip joint
|
|
Case 1
|
SCO2003P1972
|
7
|
M
|
AD
|
FBN1
|
CS II
|
Marfan Syndrome
|
Het
|
NM_000138.4
|
maternal
|
Nonsense
|
c.2649G>A p.(Trp883Ter)
|
0
|
5.33
|
119
|
|
Case 2
|
SCO1908P0067
|
18
|
M
|
AD
|
COL1A1
|
AIS
|
Osteogenesis imperfecta
|
Het
|
NM_000088.3
|
de novo
|
Stop gain
|
c.1081C>T p.(Arg361Ter)
|
0
|
3.97
|
69.9
|
bone fragility; scoliosis; osteopenia
|
|
Case 2
|
SCO1908P0067
|
18
|
M
|
AD
|
FBN1
|
AIS
|
Marfan Syndrome
|
Het
|
NM_000138.4
|
maternal
|
Missense
|
c.1453C>T p.(Arg485Cys)
|
0
|
5.33
|
119.0
|
|
Case 3
|
PCT2007P0019
|
8
|
F
|
AD
|
NF1
|
NFS
|
Neurofibromatosis, Type 1
|
Het
|
NM_000267.3
|
paternal
|
Frameshift
|
c.2307del p.(Thr770LeufsTer21)
|
0
|
5.25
|
18.24
|
cafe´-au-lait macules; scoliosis; bone fragility; blue sclera
|
|
Case 3
|
PCT2007P0019
|
8
|
F
|
AD
|
COL1A1
|
NFS
|
Osteogenesis imperfecta
|
Het
|
NM_000088.3
|
paternal
|
Splicing
|
c.2028+4 A>G
|
0
|
0
|
0
|
|
Case 4
|
WAX677
|
6
|
M
|
AD
|
ANKRD11
|
GHD
|
KBG syndrome
|
Het
|
NM_013275.5
|
de novo
|
Nonsense
|
c.4750G>T p.(Glu1584Ter)
|
0
|
5.08
|
48
|
short stature; depressed nasal bridge; long philtrum; low-set ears; tongue thrusting
|
|
Case 4
|
WAX677
|
6
|
M
|
AD
|
COL11A1
|
GHD
|
Marshall syndrome
|
Het
|
NM_001854.3
|
paternal
|
Frameshift
|
c.2508dup p.(Leu837ThrfsTer81)
|
0
|
0
|
0
|
|
Case 5
|
AX168
|
8
|
F
|
AD
|
NF1
|
ISS
|
Neurofibromatosis, Type 1
|
Het
|
NM_000267.3
|
NA
|
Splicing
|
c.6705-1 G>A
|
0
|
5.59
|
18
|
scoliosis; short stature; abnormality of the cerebral white matter; cafe-au-Lait macules
|
|
Case 5
|
AX168
|
8
|
F
|
AD
|
GLI2
|
ISS
|
Culler-Jones syndrome
|
Het
|
NM_005270.4
|
NA
|
Frameshift
|
c.1189del p.(Val397CysfsTer124)
|
0
|
0
|
0
|
|
Case 6
|
AX282
|
12
|
F
|
AD
|
TP63
|
ISS
|
Rapp-Hodgkin Syndrome
|
Het
|
NM_003722.4
|
NA
|
Nonsense
|
c.109C>T p.(Arg37Ter)
|
0
|
3.84
|
22.7
|
short stature; low set ear; low posterior hairline; scoliosis; hyperpigmentation; webbed neck
|
|
Case 6
|
AX282
|
12
|
F
|
AD
|
PTPN11
|
ISS
|
Noonan Syndrome
|
Het
|
NM_002834.3
|
NA
|
Missense
|
c.1510A>G p.(Met504Val)
|
0
|
5.13
|
25.6
|
|
Case 7
|
RDD2001P0005
|
2
|
M
|
AD
|
FBN2
|
CPC
|
Beals Syndrome
|
Het
|
NM_001999.3
|
paternal
|
Missense
|
c.3437A>G p.(Tyr1146Cys)
|
0
|
5.13
|
18.69
|
joint contractures; atrial septal defect; clinodactyly of fingers
|
|
Case 7
|
RDD2001P0005
|
2
|
M
|
AD
|
ANKRD11
|
CPC
|
KBG Syndrome
|
Het
|
NM_013275.5
|
de novo
|
Frameshift
|
c.3024_3025del p.(Lys1009GlyfsTer8)
|
0
|
0
|
0
|
|
Case 8
|
P19009402
|
4
|
M
|
AD
|
FGFR2
|
Syndactyly
|
Apert Syndrome
|
Het
|
NM_000141.4
|
de novo
|
Missense
|
c.755C>G p. (Ser252Trp)
|
0
|
5.79
|
23.5
|
cloverleaf skull; orbital hypertelorism; proptosis; midfacial hypoplasia; syndactyly of the hands and feet; hyperthermia
|
|
Case 8
|
P19009402
|
4
|
M
|
AD
|
RYR1
|
Syndactyly
|
Malignant Hyperthermia Susceptibility 1
|
Het
|
NM_000540.2
|
paternal
|
Frameshift
|
c.12788_12793dup p.(Glu4263_Gly4264dup)
|
3.36×10-5
|
0
|
0
|
{kind=link}