Patient samples. In the present study, 40 bladder cancer tissues and 40 adjacent non-tumor tissues were collected from Meizhou People’s Hospital. Patients had not received radiation therapy or chemotherapy prior to surgery treatment. The clinical stage of patients with bladder cancer was determined using the World Health Organization criteria. Tumor tissues were stored in liquid nitrogen or at -80˚C. The present study was approved by The Institutional Review Board of Meizhou People’s Hospital. Written informed consent was obtained from all patients.
Cell culture and transfection. A normal bladder cell line (SV-HUC-1) and bladder cancer cell lines (5637, J82, ECV-304, BIU-87, T24, HCV29 and H/RB-CL2) were purchased from American Type Culture Collection. Cells were cultured in RPMI-1640 (Invitrogen; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS, 2 mM glutamine and 100 µg/ml streptomycin/penicillin (Sangon Biotech Co., Ltd.) at 37˚C with 5% CO2.
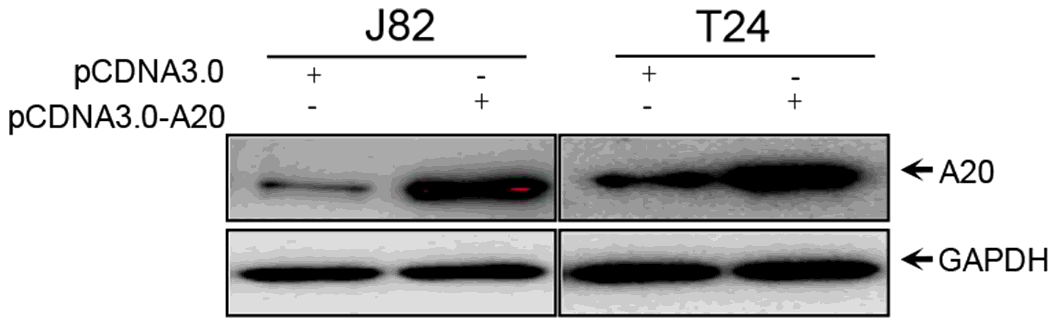
Western blotting. Western blotting was performed as previously described. Briefly, total protein was extracted using NP40 buffer (Beijing Solarbio Science & Technology Co., Ltd.) at 4˚C for 20 min, followed by centrifugation at 12,000 x g at 4˚C for 10 min. Proteins were separated via 12% SDS-PAGE and transferred to PVDF membranes (Beijing Solarbio Science & Technology Co., Ltd.). Following blocking with 5% BSA in PBST (0.1% Tween 20) at room temperature for 1 h, the membranes were incubated with primary antibodies at 4˚C overnight. Subsequently, the membranes were incubated with a horseradish peroxidase-conjugated secondary antibody at 37˚C for 1 h. Protein bands were visualized using ECL reagent. GAPDH was used as the loading control.
Reverse transcription-quantitative PCR (RT-qPCR). Total RNA was extracted using TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer’s protocol. Total RNA (1 mg) was reverse transcribed into cDNA using the PrimeScript™ RT reagent kit with gDNA Eraser (Takara Biotechnology Co., Ltd.). Subsequently, qPCR was performed using an RT-qPCR machine (Applied Biosystems; Thermo Fisher Scientific, Inc.). miRNA and mRNA expression levels were normalized to the internal reference genes U6 and GAPDH, respectively.
Apoptosis analysis and Cell cycle analysis. Collected different groups of cells, each group has no less than 1×105 cells. Wash the cells with PBS twice. According to the instructions of the apoptotic kit (KeyGEN Biotech China), add the dyes in turn, protect from light for 10min, flow Instrument (ACEA Biosciences, USA) detection and analysis. Collect different groups of cells, and wash the cells with PBS twice. Then cells were fixed by 70% ethanol overnight. According to the cell cycle kit (KeyGEN Biotech, China), dyes were added sequentially, protected from light for 30min, and analyzed by flow cytometry(ACEA Biosciences, USA).
Cell Counting Kit-8 (CCK-8) assay. Cells (1x104 cells/well) were seeded into 96-well plates. Following culture for 48 or 72 h, cell proliferation was analyzed by performing the CCK-8 assay (Dojindo Molecular Technologies, Inc.) according to the manufacturer’s protocol. Absorbance was measured at a wavelength of 450 nm using a Multiskan™ GO Microplate Spectrophotometer (Thermo Fisher Scientific, Inc.).
Cell migration assay. Cell migration was measured using 6.5-mm Transwell inserts with 8.0-mm pore polycarbonate membranes (Costar; Corning Inc.). Cell invasion was assessed using 6.5-mm Transwell inserts with 8.0-mm pore polycarbonate membranes (Costar; Corning Inc.). Cell migration and invasion were determined as previously described. Subsequently, the average number of migratory/invading cells was counted.
Wound healing assay. At 24 or 36 h post-transfection, cells were harvested and cultured for a further 24 h. Subsequently, a single scratch in the cell monolayer was made using a 300-µl sterile pipette. The wounds were observed at 24 h. The intersection of the bottom line and the cell scratch line was considered as the observation point.
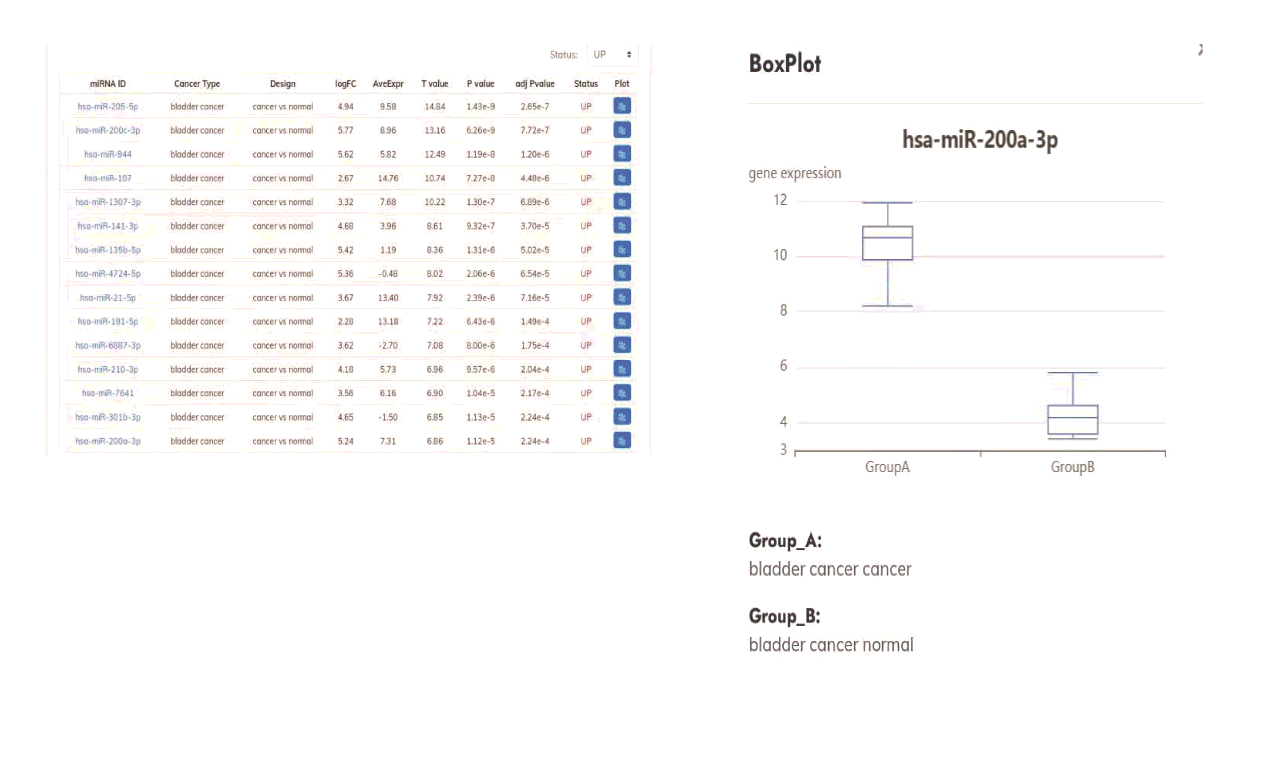
Dual-luciferase reporter assay. The wild-type (WT) A20 3’UTR sequence was amplified via PCR and cloned into the pmirGLO vector (Promega Corporation). To construct the mutant (Mut) plasmid, the complementary sequences for miR-200a-3p in the 3’UTR of A20 were mutated. J82 and T24 cells were co-transfected with A20-WT or A20-Mut and miR-200a-3p mimic or miR-NC. At 36h post-transfection, luciferase activities were measured using the DLR dual luciferase reporter assay system (Promega Corporation).
Tumor growth in vivo. The function of miR-200a-3p in bladder cancer growth was assessed by evaluating tumor growth in vivo. Male nude mice (age, 6 weeks; n = 6 per group) were used in the present study to assess tumor growth and metastasis. To assess tumor growth, mice were subcutaneously injected with miR-200a-3p-overexpression or control cells (1x105 ). Tumor volume was calculated at 1, 2, 3 and 4 weeks post-injection. At 4 weeks post-injection, all mice were euthanized and the tumors were isolated. All animal experiments were approved by the ethics committee of Meizhou People’s Hospital
Western blotting. Proteins were separated via 12% SDS-PAGE and transferred onto PVDF membranes (EMD Millipore). Following blocking with 5% BSA and washing three times with PBST (1% Tween-20) the membranes were incubated with primary antibodies. Subsequently, the membranes were incubated with a HRP-conjugated goat anti-mouse IgG secondary antibody (Sigma-Aldrich; Merck KGaA). Protein bands were visualized using ECL detection reagents (cat. no. E412-02; Vazyme Biotech Co., Ltd.) and scanned using the ChemiDoc XRS + Imaging System (Bio-Rad Laboratories, Inc.).
Statistical analysis. Data are presented as the mean ± SD from at least three independent repeats. Statistical analyses were performed using SPSS software (version 19.0; IBM Corp.). Comparisons among multiple groups were analyzed using one-way or two-way ANOVA followed by Bonferroni’s post hoc test. Comparisons between two groups were analyzed using a paired or unpaired Student’s t-test. P < 0.05 was considered to indicate a statistically significant difference.
{kind=link}
{kind=link}