Reagents
High and low molecular weight hyaluronic acid (HA) were provided by Altergon (Altergon s.r.l., Italy). These are fermentative HA of high purity derived from Streptococcus equi ssp. equi, extensively purified at pharmaceutical grade (e.g., purity >95%, water content <10%, EU/mg <0.05, and very low metal contents). The raw materials were fully characterized through hydrodynamic analyses using Size-exclusion chromatography coupled to a triple detector (SEC-TDA, Viscotek Malvern) [13]. Hybrid cooperative complexes of hyaluronic acid (HCC), obtained through a thermal procedure for the formation of hybrid cooperative complexes of hyaluronic acid, starting from an initial mixture of an equal amount (ratio 1:1) of HHA (Mw = 1400±200 kDa; Mw/Mn = 1.4) and LHA (Mw = 100±20kDa; Mw/Mn = 1.4). The starting concentration used was 32g/L: 16 mg HHA + 16 mg LHA in 1 mL volume. The final concentration used in these experiments was 1.6 mg/mL obtained by opportunely diluting all solutions with the culture medium (DMEM Dulbecco’s modified Eagle’s medium, Gibco, Invitrogen, and/or OM osteogenic medium).
Dental pulp extraction and culture
All experimental procedures involved were approved by the Ethics Committee of University of Campania approved on June 12th, 2005, Internal Registry: Experimentation #914 and were performed in line with the principles of the Declaration of Helsinki.
Human dental pulps were extracted from teeth of healthy adults (aged 21–38 years) as described previously [7]. All participants signed the Ethical Committee (University of Campania Internal Ethical Committee) consent form. Every participant was pretreated for a week with professional dental hygiene. The dental crown was covered with 0.3% chlorhexidine gel (Forhans) for 2 min before the extraction. Dental pulp was obtained with a dentinal excavator or a Gracey curette. The pulp was delicately removed and immersed for 1 h at 37 °C in a digestive solution composed of 3 mg/ml of type I collagenase and 4 mg/ml of dispase in PBS containing 40 mg/ml of gentamicin. Once digested, the solution was filtered through 70 μm Falcon strainers (Becton & Dickinson). Cells were cultured in basal growth medium (standard medium) consisting of Dulbecco's modified Eagle's medium (DMEM) with 100 units/ml of penicillin, 100 mg/ml of streptomycin, and 200 mM l-glutamine (all from Gibco), supplemented with10% heat-inactivated AB-HS (Invitrogen). Cells were maintained in a humidified atmosphere under 5% CO2 at 37◦C and the media were changed twice a week.
hDPSCs isolation and osteogenic differentiation
At the first passage of culture, cells were detached using trypsin–EDTA (GIBCO). At least 200,000 cells were incubated with fluorescent-conjugated antibodies for 30 min at 4◦C, washed, and resuspended in PBS. The antibodies used in this study were: anti-CD34 PE (BD Pharmingen, Buccinasco, Milano, Italy), anti-CD90 FITC (BD Pharmingen, Buccinasco, Milano, Italy) and anti-CD45 APC-Cy7 (BD Pharmingen, Buccinasco, Milano, Italy). Isotypes were used as controls. Cells were analyzed with FACS ARIA III (BD Biosciences, San Jose, CA, USA) and data collected with Diva Software. Cells were sorted using simultaneous positivity for CD90 and CD34 using a FACS ARIA III (BD, Franklin Lakes, NJ, USA) as previously reported [7]. The purity of sorted populations was routinely 90%.
hDPSCs were treated with HHA, LHA, HCC diluted in DMEM and osteogenic media (OM). All experiments were performed at 7, 14 and 21 days. Media were changed twice a week. Cells grown in DMEM or OM without hyaluronans were used as controls.
For osteogenic differentiation, hDPSCs were cultured in osteogenic medium containing 100 nmol/L dexamethasone, 10 mmol/L beta-glycerophosphate, and 0.05 mmol/L L-ascorbic acid-2-phosphate. Osteogenic differentiation was evaluated by Alizarin Red S (ARS) and the expression of bone-related markers such as osteocalcin (OC), osteopontin (OPN), and bone sialoprotein (BSP) at 7, 14 and 21 days. For Alizarin Red S staining, samples were washed twice in PBS, fixed with 4% paraformaldehyde (PFA) for 30 min at 4°C, and stained with 2% Alizarin Red solution, pH 4.2 (Sigma Aldrich, Milan, Italy) for 20 min at room temperature. Stained cells were extensively washed with deionized water to remove any non-specific precipitation. Micrographs were taken using a microscope Eclipse TE2000-S (Nikon) and a Nikon camera. The number of bone nodules and area calculation of positive staining were measured and compared amongst groups. Quantification of the staining intensity was measured using ImageJ software (National Institutes of Health (NIH), Bethesda, MD, United States).
YAP/TAZ inhibitor-1 treatment and cell viability assay
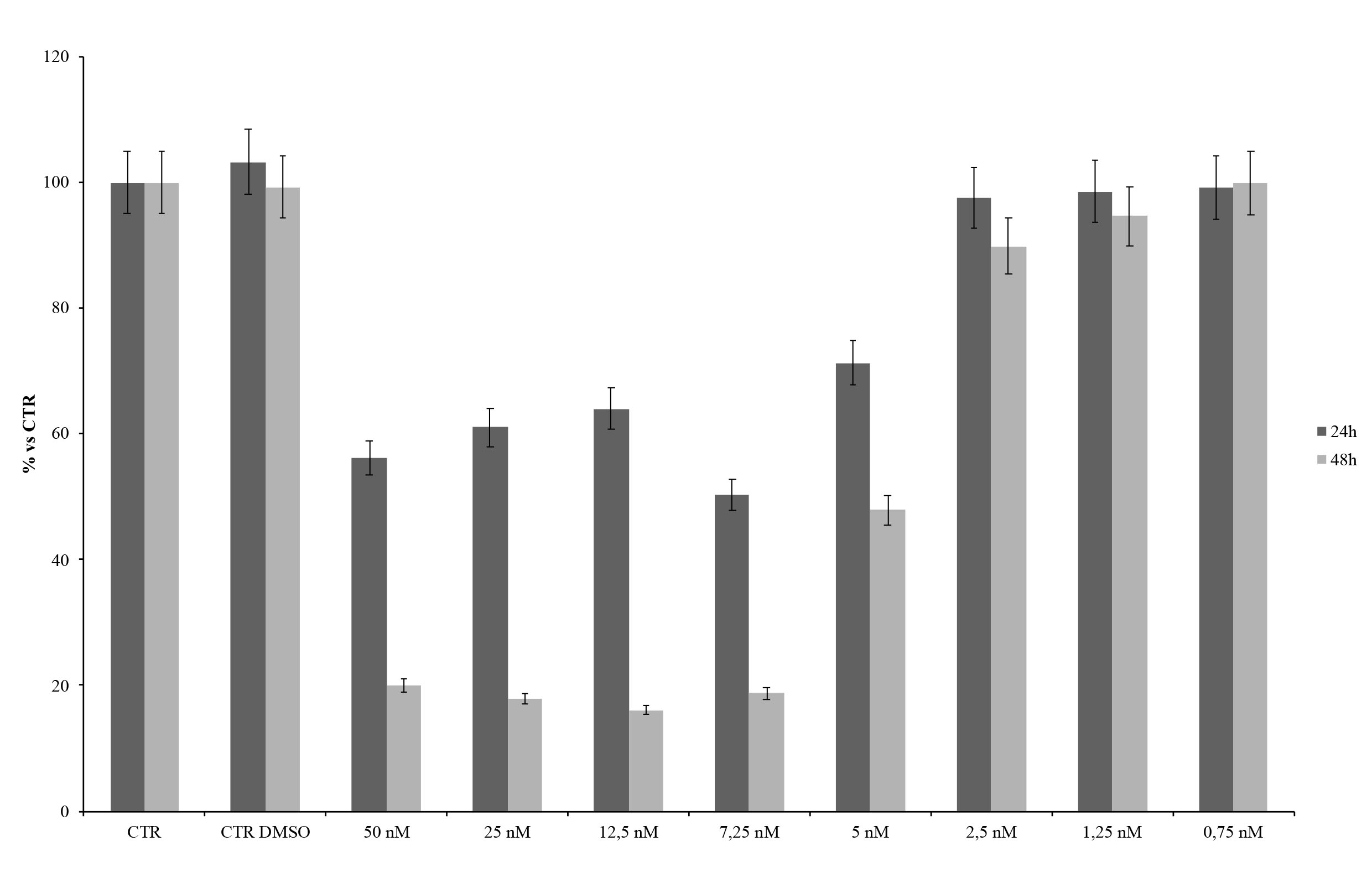
To confirm involvement of YAP/TAZ pathway in promoting osteogenic differentiation, hDPSCs were treated with hyaluronans and YAP/TAZ inhibitor-1 (MedChemExpress.). To evaluate the cytoxicity of YAP/TAZ inhibitor-1, hDPSCs viability was measured by the colorimetric 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were seeded in 96-well plates at a density of 104 cells per well, treated with different concentrations (50, 25, 12.5, 7.25, 5, 2.5, 1.25, 0.75 nM) for 24 and 48 hours. After incubation, they were treated with 100 μL of 1 mg/mL MTT (Sigma) in DMEM medium containing 10% FBS for 4 h at 37 °C. The medium was then replaced with 200 μL of DMSO and shaken for 15 min. Absorbance at 540 nm was measured using a microplate ELISA reader with DMSO used as the blank.
YAP/TAZ inhibitor-1 treated hDPSCs were cultured in DMEM at 10% FBS for 24, 72 hours and 7 days. YAP, TAZ, RUNX-2, OC, OPN, and BSP gene expression were evaluated by real Time PCR. OC, OPN, and BSP protein levels were checked also by western blotting.
RNA extraction and quantitative real-time PCR analyses
hDPSCs treated with HHA, LHA, and HCC (0.16% w/v) for 7, 14, and 21 days, and with YAP/TAZ inhibitor-1 for 24, 72 hours and 7 days, were directly lysed with TRIzol® (Invitrogen, Milan, Italy). Following precipitation with isopropyl alcohol and washing with 75% ethanol, the RNA pellets were re-suspended in nuclease-free water. The concentration of the extracted RNA was determined through a Nanodrop spectrophotometer (Celbio, Milan, Italy) and 1µg of DNase-digested total RNA was retro-transcripted in the cDNA using Reverse Transcription System Kit (Promega, Milan, Italy). Quantitative real-time PCR was obtained by iQTM SYBR® Green Supermix (BioRad Laboratories Srl, Milan, Italy) to analyze the gene expression of some osteogenic biomarkers such as Osteocalcin (OC), Osteopontin (OPN) and Bone sialoprotein (BSP). Also, Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ), were analyzed together with the two YAP/TAZ regulated genes, namely the connective tissue growth factor (CTGF) and ankyrin repeat domain-containing protein 1 (ANKRD-1). Finally, runt domain transcription factors (RUNX-1 and 2) were evaluated.
Oligonucleotide sequences used for quantitative PCR are reported in Table 1 and were obtained by Beacon DesignerTM software (BioRad Laboratories Srl, Milan, Italy). Samples were analyzed in triplicate and the expression of specific mRNA relative to the control was determined after normalization with HPRT housekeeping gene (internal control). The fold-change of mRNA expression of the genes under evaluation was calculated by using the 2−∆∆Ct comparative threshold method (∆Ct = difference of ∆Ct between treated cells and untreated cells used as controls). The results were expressed as normalized fold expression, calculated by the ratio of crossing points of amplification curves of several genes and internal standard, by using the Bio-Rad iQTM5 software (Bio-Rad Laboratories Srl) as previously reported [1].
Protein levels evaluation of OC, OPN, and BSP by western blotting analyses
Following HA-based gels and YAP/TAZ inhibitor-1 treatments, hDPSCs were lysed by a Radio-Immunoprecipitation Assay (RIPA buffer) (1x) (Cell Signaling Technology), protein concentrations were evaluated using the Bradford method, and the western blotting analyses were performed according to previously described protocols [1]. In particular, 30 μg of intracellular proteins were electrophoretically resolved on 15% SDS-PAGE and transferred to a nitrocellulose membrane (GE, Amersham, UK). This latter was blocked with 5% nonfat milk in Tris-buffered saline and 0.05% Tween-20 (TBST) and primary antibodies to detect OC (Abcam, Cambridge, UK), OPN (Abcam, Cambridge, UK) and BSP (Abcam, Cambridge, UK) were used at 1:500 dilutions and incubated for 2 h at room temperature (RT). The nitrocellulose membrane was extensively washed with TBST and immunoreactive bands were detected by chemiluminescence using corresponding horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology, CAUSA), diluted 1:10000 for 1 h, at RT and reacted with an ECL system (Millipore). Protein levels were normalized versus the signal obtained with an anti-Actin antibody 1:500 dilutions (Santa Cruz Biotechnology, CA, USA) and anti-GAPDH antibody diluted 1:500 (Sigma). The semi-quantitative analysis of protein levels was carried out using the Gel Doc 2000 UV System according to the manufacturer’s protocol.
CD44 expression in hDPSCs by immunofluorescence staining
Monolayers of hDPSCs were cultured and treated with HA-based gels in standard and osteogenic media in four-well covered glass chamber slides. After 48 h of treatment, the cells were fixed with 4% w/v paraformaldehyde in phosphate-buffered saline (PBS) for 15 min at room temperature and permeabilized in 0.2% Triton X-100 v/v in PBS for 1 h. Non-specific sites were blocked using blocking buffer solution (PBS containing 10% v/v bovine serum and 1% w/v BSA). The cells were then incubated with anti-CD44 antibody (Cell signaling, Leiden, The Netherlands) overnight at 4°C followed by incubation with corresponding secondary antibody for 2 h at room temperature. Nuclei were stained with 20-(4-hydroxyphenyl)-5-(4-methyl-1-piperazinyl)-2.50-bi-1H-benzimidazole trihydrochloride hydrate, bisBenzimide (Hoechst) and actin filaments were stained using phalloidin tetramethylrhodamine B isothiocyanate. (Sigma-Aldrich, Milan, Italy). Fluorescence images were captured using a fluorescence microscopy system (Nikon, Tokyo Japan).
YAP-TAZ expression in hDPSCs by immunofluorescence staining
hDPSCs were cultured with HA-based gels in standard medium and treated with YAP/TAZ inhibitor-1 in 24-well covered glass chamber slides. After 72h of treatment, the cells were fixed with 4% w/v paraformaldehyde in phosphate-buffered saline (PBS) for 15 min at room temperature and permeabilized in 0.2% Triton X-100 v/v in PBS for 1 h. Non-specific sites were blocked using blocking buffer solution (PBS containing 10% v/v bovine serum and 1% w/v BSA). The cells were then incubated with anti-YAP and anti-TAZ antibodies (Invitrogen) overnight at 4°C followed by incubation with corresponding secondary antibody for 2 h at room temperature. Nuclei were counterstained with 20-(4-hydroxyphenyl)-5-(4-methyl-1-piperazinyl)-2.50-bi-1H-benzimidazole trihydrochloride hydrate, bisBenzimide (Hoechst). Cells were imaged with a fluorescence microscope EVOS FL Cell Imaging System (Thermo Scientific, Rockford, USA).
Cell Seeding and Differentiation in 3D
To achieve 3D tissue formation, hDPSCs were seeded on a Gingistat (GABA VEBAS, Roma, Italy, http://www.gaba-info.it/) scaffold. This scaffold is a lyophilized collagen type I sponge. Collagen sponges were cut under sterile conditions into 5×5×5 mm3 cubes. Scaffold cubes were placed in six-well plates and a cell suspension of 1 × 106 cells contained in 100 µl medium was pipetted onto the top of each cube. Cells were allowed to adhere under a humidified atmosphere at 37°C and 5% CO2 for 4 hours. The seeded scaffolds were then placed in the osteogenic medium with HA-based gels and cultured for 21 days in an incubator at 37°C and 5% CO2. Media were changed twice a week.
After 3D culture, samples were fixed in 4% paraformaldehyde (PFA) and cryoprotected overnight at 4°C by immersion in a 30% (wt/vol) sucrose solution before being embedded in Tissue-Tek® O.C.T. Compound (Tissue-Tek; Sakura Finetek, Torrance, CA, http://www.sakuraus.com.) and frozen.
Osteogenic differentiation of frozen sections was evaluated by Alizarin Red S staining as described previously. Briefly, the samples were washed in highly purified H2O to remove O.C.T. for 5 min and then stained with Alizarin Red solution (2%, pH 4.2; Sigma Aldrich, Milan, Italy) for 20 min at room temperature. Stained samples were extensively washed with deionized water to remove any nonspecific precipitation. Micrographs were taken with a microscope Eclipse TE2000-S (Nikon, Firenze, Italy) and a Nikon camera (Nikon, Firenze, Italy).
For immunofluorescence staining, frozen sections were permeabilized with 0.1% Triton X-100 for 15 min. Each sample was incubated in PBS containing 5% bovine serum albumin (BSA) for 30 min. at room temperature as a blocking step. Then, after washing twice with PBS, samples were incubated with primary antibodies: mouse monoclonal to OC (1:100, Santa Cruz, Heidelberg, Germany), rabbit polyclonal to OPN (1:1000, Abcam, Cambridge, UK), overnight at 4 °C in the dark. This step was followed by incubation with the secondary antibody Tetramethylrhodamine (TRITC)-conjugates (1:1000, Abcam). Nuclear counterstaining was performed with 4,6-diamidino-2-phenylindole (DAPI). After extensive washing with PBS, coverslips were mounted onto slides. Images were collected under a fluorescence microscope (EVOS FL Cell Imaging System, Thermo Scientific, Rockford, USA).
Statistical analysis
Values are shown as the mean ± SD of measurements of at least three independently performed experiments to avoid possible variation of cell cultures. Student's t-test was employed, and p<0.05 was considered to be statistically significant.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}