1. Identification of genome instability-associated lncRNAs in patients with pancreatic cancer
To detect the potential Genome instability-related lncRNAs, the cumulative number of somatic mutations in each patient with pancreatic cancer was calculated from TCGA. The first 25% (n =43) and the last 25% (n =40) patients were classified into GU group and GS group by the descending order of cumulative number. Then the lncRNA expression profiles in GU group and GS group were analyzed by unsupervised clustering, the result show that total 206 lncRNAs were found to be significantly differentially expressed (Figure 2A). All patients with pancreatic cancer in TCGA were divided into GU-like group and GS-like group by unsupervised hierarchical clustering analysis based on the expression levels of the 206 differentially expressed lncRNAs. The cumulative number of somatic mutations was higher in GU-like group and lower in GS-like group (Figure 2B). As shown in Figure 2C, more mutated genes exist in GU-like group(P<0.001,Mann-Whitney U test). As UBQLN4 gene is one of the driving factors of gene instability, the expression level of UBQLN4 gene in GU-like group and GS-like group was compared. The results showed that there was significant difference in the expression level of UBQLN4 between the two groups, and the expression level of UBQLN4 in GU-like group was significantly higher than that in GS like group. (P < 0.001, Mann–Whitney U test, Figure 2D).
To better understand the biological significance of the 206 differentially expressed lncRNAs,functional enrichment analysis was performed to predict potential functions. We selected the protein coding genes (PCGs) most related to the expression of each lncRNAs to construct an lncRNA-mRNA co-expression network (Figure 3A). According to the enriched results of the lncRNA-correlated PCGs, GO biological process (e.g., cellular component (CC), DNA binding in the molecular function (MF), and metabolism in the biological process (BP)) and KEGG pathway (e.g., MAPK signaling pathway, cAMP signaling pathway, Pancreatic secretion and Endocrine resistance) were annotated to be associated with genome instability(Figure 3B-C). Based on the above results, it is considered that the 206 lncRNAs were involved in the genomic instability-related biological process, and their altered expression may destruct the genomic stability of cells. Therefore,the 206 differentially expressed lncRNAs were recognized as candidate lncRNAs with genomic instability in pancreatic cancer.
2. Acquisition of a genomic instability-associated lncRNA prognostic signature from the training set
In order to screen out the prognostic lncRNAs with independent value, we performed univariate Cox proportional hazard regression analysis to analyze the relationship between expression levels of 206 GIlncRNA and OS in the training set, 17 candidate prognostic lncRNAs were found to be significantly associated with the prognosis of pancreatic cancer patients (Figure 4A). Furthermore, multivariate Cox proportional hazards regression was used to analysis on 17 candidate prognostic lncRNAs. Based on the multiCox model (Figure 4B), 5 of 17 candidate lncRNAs including AL121772.1, BX640514.2, LINC01133, AC087752.3 and LYPLAL1-AS1 were found to retain their prognostic significance and thus were identified as independent prognostic lncRNAs (P<0.05). Among five prognostic lncRNAs, one lncRNAs (AC087752.3) having negative coefficients was shown to be a protective factor whose high expression level was closed associated with a longer survival, whereas the remaining four lncRNAs (AL121772.1, BX640514.2, LINC01133 and LYPLAL1-AS1) with positive coefficients tended to be prognostic risk factors and their high expression were associated with a shorter survival. A risk score model of genomic instability-associated lncRNA signature (GILncSig) based on the expression level and multivariate Cox analysis regression coefficients of 5 independent prognostic lncRNAs was generated to predict the outcome of pancreatic cancer patients as follows:GILncSig = (-1.61 × expression value of AC087752.3) + (0.63 × expression value of AL121772.1) + (0.39× expression value of BX640514.2) + (0.02 × expression value of LINC01133) + (0.38× expression value of LYPLAL1-AS1). According to the GILncSig model, the prognostic risk score was computed for each patient in the training set. Using the median risk score as the cutoff point, all patients of training set were classified into a high-risk group (n = 38) and a low-risk group (n = 46). The Kaplan-Meier analysis indicated that the overall survival was significantly different between the two risk groups and patients in low-risk subgroup had markedly longer overall survival than those in the high-risk group (P=0.009, log-rank test, Figure 5A). The time-dependent receiver operating characteristic (ROC) curves analysis for GIlncRNA prognostic model achieved an area under the curve (AUC) of 0.653 at 1 year of overall survival (Figure 5C). These results demonstrated the GIlncRNA had better prognosis prediction performance in patients with pancreatic cancer. Then we ranked the risk scores of patients in the training set. Figure 5B showed the expression pattern of the 5 Independent prognostic lncRNAs, the expression level of UBQLN4 and the count of somatic mutations. We found that for patients with high-risk scores, the expression levels of four risk lncRNAs(AL121772.1、BX640514.2、LINC01133、LYPLAL1-AS1) were up-regulated, while one protective lncRNA(AC087752.3) was expressed at a low level. In contrast, these prognostic lncRNAs expressed the opposite patterns in patients with low-risk scores. Similarly, there were significant differences in UBQLN4 expression level between high-risk group and low-risk group (P=0.049, Mann–Whitney U test; Figure 5D). Moreover, Figure 5D also revealed that the number of somatic mutations in high-risk group were slightly higher than those in low-risk group (P=0.09, Mann–Whitney U test; Figure 5D).
3.Validation of GILncSig in the testing set and entire TCGA set
To confirm our findings, the prognostic performance of the GILncSig was further evaluated in the testing set. Patients in the testing set were divided into the high-risk group (n = 43) and the low-risk group (n = 44) by using the same GILncSig and cutoff value deriving from the training set. Kaplan-Meier curves showed that there was a significant difference in overall survival between the high-risk group and the low-risk group, and the overall survival of the high-risk group was much lower than the low-risk group (p<0.001, log-rank test, Figure 5E), which were similar to those observed in the training set. Validation of the GILncSig in the testing set of 87 patients produced an ROC with an AUC of 0.806 at 1 year (Figure 5G). Figure 5F shows how the expression level of GILncSig, the count of somatic mutation, and the expression level of UBQLN4 in the testing set change with the increasing score. The analysis indicated that Somatic mutation counts and the expression level of UBQLN4 were significantly higher in the high-risk group as compared with those in the low-risk group (P=0.0044,P=0.00054, Mann-Whitney U test; Figure5H).
Similar results were observed when the prognostic performance of the GILncSig was further used to the entire TCGA set. Like the training and testing set, the GIlncRNA was able to stratify 171 pancreatic cancer patients of the entire TCGA set into the high-risk group (n=81) and low-risk group (n=90) with obviously different overall survival (P<0.001, log-rank test, Figure 6A). The AUC of time-dependent ROC analysis for overall survival in the entire TCGA set was 0.724 (Figure 6B). The expression of GILncSig, somatic mutation counts and UBQLN4 expression level of pancreatic cancer patients in TCGA set were presented in Figure 6C, which were similar to those observed in the training set and testing set. The counts of somatic mutations in high-risk group were significantly higher than that in low-risk group (P=0.0022, Mann-Whitney U test, Figure 6D), as was the expression level of UBQLN4 (P=0.0001, Mann-Whitney U test, Figure 6D).
4.Comparison of the GILncSig and other lncRNA-related predictive signatures for survival prediction
Recently, two lncRNA-related signatures were reported to predict prognosis of pancreatic cancer patients. Therefore, we further compared the prognostic value of our GILncSig to that of different lncRNA-associated signatures for predicting outcomes: the five-lncRNA signature derived from Song’s study (hereinafter referred to as SongSig)[26] and the three-lncRNA signature derived from Shi’s study (hereinafter referred to as ShiSig)[27]. Utilizing the same TCGA patient set. Then we performed the time-dependent ROC analysis and calculated the area under the ROC curves to compare the prediction performance between the GILncSig and other two existing lncRNA-related signatures in the entire TCGA set. The result demonstrated that the AUC at1 year of overall survival for the GILncSig is 0.724, which was significantly higher than that of SongSig (AUC = 0.642) and ShiSig (AUC = 0.556) (Figure 7). For this reason, we believed that the GILncSig had better prognostic power than those two lncRNA-related signatures.
5. Independence of prognostic value of the GIlncRNA from other clinical variables
To determine whether the prognostic value of the GIlncRNA was independent of other clinical variables. Multivariate Cox regression analysis was performed in each patient set using prognostic risk score, age, gender, pathological grade and stage. Results from multivariate Cox analysis revealed that the GIlncRNA was significantly associated with overall survival in each set when adjusted for age, gender, pathological grade and stage (Table 1). At the same time, we also observed that age, gender, pathological grade and stage were different in the multivariate analysis significantly. So we further performed data stratification analysis according to age and gender, pathological grade and stage. According to the age, pancreatic cancer patients could be stratified into an old patient group (age > 65, n=81) and a young patient group (age ≤65, n=90). The GIlncRNA could subdivide each age group into high-risk group and low-risk group. There was significantly different overall survival between high-risk group and low-risk group in each age group. (log-rank test p=0.016 for the old patient group and log-rank test p<0.001 for the young patient group) (Figure 8A). Next, all patients were also stratified by gender. 78 female patients were classified into high-risk group (n =37) and low-risk group (n = 41) based on the GIlncRNA. Similarly, 93 male patients were separated into two groups (high-risk group: n=44; low-risk group: n=49). The overall survival of patients in the low-risk group was significantly longer than that of patients in the high-risk group by analysis of the results. (log-rank test p=0.002 for the female group; log-rank test p=0.001 for the male group; Figure 8B). In addition, all patients in entire TCGA set were grouped according to tumor size, lymph node metastasis and distant metastasis. Each group was further separated into high-risk group and low-risk group by the GIlncRNA, and the difference of overall survival between the two groups was compared. As shown in Figure 8, with the exception of the metastatic group (M1 group), there were statistically significant differences in overall survival between the high-risk and low-risk groups in each group, whereas marginally significant difference was existed in high-risk subgroup and low-risk subgroup of M1 group (p=0.052 for T1-2 group, p<0.001 for T3-4 group, Figure 8D; p=0.027 for N0 group, p=0.03 for N1 group, Figure 8G; p=0.009 for M0 group, p=0.317 for M1 group, Figure 8C; log-rank test). Finally, the same analysis method was applied to the pathological grade and stage of patients. All patients were divided into low-grade (G1/G2) and high-grade groups (G3/G4) according to pathological grade. The results of stratified analysis showed that the patients with high-grade were divided into either a high-risk group (n=24) with shorter survival or a low-risk group (n=25) with longer survival (p=0.068, log-rank test; Figure 8E). The patients in the low-grade group were similarly classified into two risk subgroups with significantly different survival time (p<0.001, log-rank test; Figure 8E). Furthermore, patients with pathologic stage I or II were combined into an early-stage group (n=161) and that with pathologic stage III or IV were combined into a late-stage group (n=7). The GIlncRNA divided the early-stage group and the late-stage group into high-risk group and low-risk group respectively. The overall survival was significantly different between the two groups among the early-stage group (p<0.001, log-rank test; Figure 8F). Nevertheless, the difference in overall survival between the two groups was not significant probably due to the limited sample size in the late group (p=0.549, log-rank test; Figure 8F). Taken together, these results indicated that the GILncSig was an independent prognostic factor associated with overall survival in pancreatic cancer patients.
6.The prognostic significance of GILncSig better than KRAS, TP53, SMAD4 mutation status
KRAS, TP53 and SMAD4 were the most frequent mutant genes and associated with poor prognosis in pancreatic cancer. With this in mind, these three genes were included in the training set, testing set and TCGA set for analysis, respectively. Then further stratified analysis was performed based on the mutation status of KRAS, TP53 and SMAD4 by GILncSig. The analysis showed that the proportion of patients with KRAS, TP53 and SMAD4 mutations in high-risk group was higher than that in low-risk group to varying degrees in each set. For KRAS, 66% of the high-risk group had KRAS mutations, significantly higher than 16% of the low-risk group in the training set (chi-square test P<0.001). In the testing set, 72% of the high-risk group had KRAS mutation, which was significantly higher than 46% of the low-risk group (chi-square test P=0.040). In the entire TCGA set, 69% of KRAS mutation in high-risk group significantly higher than 33% of low-risk group (chi square test P < 0.001). These results suggest that GILncSig is closely related to the mutation state of KRAS gene. Therefore, we applied GILncSig to patients with KRAS Wild type (KRAS Wild) and KRAS mutation type (KRAS mutation). Patients with KRAS Wild were divided into low-risk group (KRAS Wild/GS-like) and high-risk group (KRAS Wild/ GU-like), and patients with KRAS mutation were divided into low-risk group (KRAS Wild/GS-like) and high-risk group (KRAS mutation/GU-like). Through comparative analysis, we found that overall survival of KRAS Wild/GS-like group was significantly different from that of KRAS Wild/ GU-like group and KRAS Wild/ GU-like group and patients in KRAS Wild/GS-like group had better prognosis (p=0.01, log-rank test; Figure 9A). For TP53, as shown in Figure 9B, 73% of TP53 mutations in the high-risk group were significantly higher than 26% in the low-risk group in the training set (chi square test P < 0.001). Similarly, in the TCGA set, the TP53 mutation in high-risk group was obviously higher than that in low-risk group (high-risk group 66% versus low-risk group 41%, chi square test P=0.004). However, TP53 mutations were only slightly higher in the high-risk group than that in the low-risk group in the test set, and there was no significant difference between the two groups (high-risk group 58% versus low-risk group 54%, chi square test P=0.874). In consequence, we believe that TP53 status can be predicted according to the GILncSig risk score. Then patients with TP53 mutation and TP53 wild type were further divided into TP53 mutation high-risk group (TP53 mutation/GU-like), TP53 mutation low-risk group (TP53 mutation/GS-like), TP53 wild high-risk group (TP53 wild/GU-like) and TP53 wild low-risk group (TP53 wild/GS-like). Survival analysis showed that patients in the TP53 wild/GS-like group had longer survival than those in the TP53 wild/GU-like group, and the higher risk scores were associated with lower survival rates in TP53 wild subgroups (p=0.002, log-rank test; Figure 9B). For SMAD4, it has similar results with KRAS and TP53. The patients in training set, testing set and TCGA set were respectively divided into high-risk group and low-risk group by using GILncSig. In each set, the proportion of SMAD4 mutation in high-risk group was significantly higher than that in low-risk group (p=0.228 for training set; p=0.028 for testing set; p=0.009 for TCGA set; chi-square test; Figure 9C). The patients with SMAD4 mutation type and SMAD4 wild type were further separated into SMAD4 mutation/GU-like group, SMAD4 mutation/GS-like, SMAD4 wild/GU-like group and SMAD4 wild/GS-like group. The results of survival analysis showed that the overall survival among the groups was slightly different (p=0.062, log-rank test; Figure 9C). Therefore, the above findings suggested that the GILncSig is superior to KRAS, TP53 and SMAD4 mutation status in prognosis.
7. Development and validation of a nomogram for predicting survival in patients with pancreatic cancer
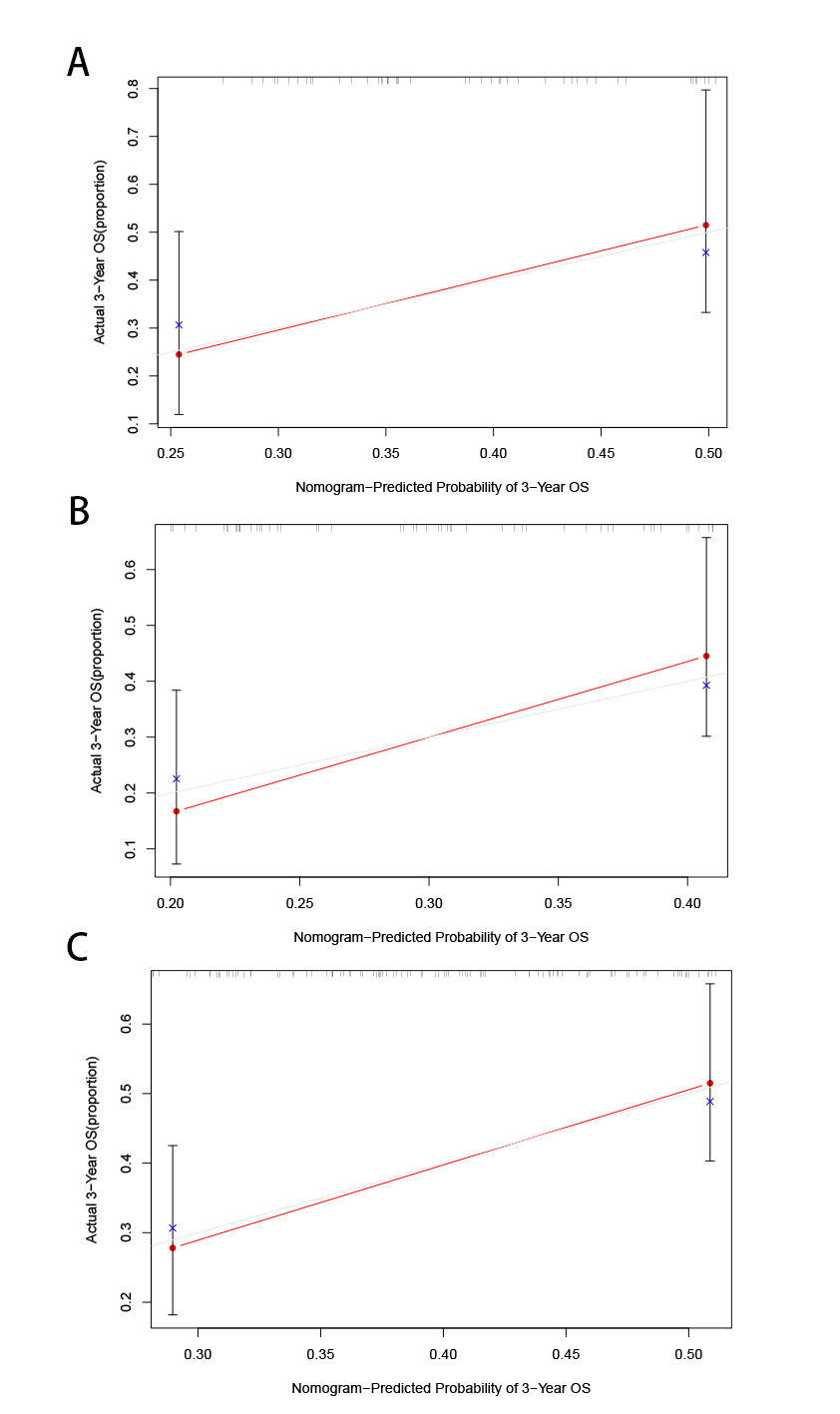
In order to improve the clinical application of the GILncSig, we established a prognostic nomogram model combined with risk score, age, gender, pathological grade and stage to predict the patients’ survival at 1-, 2-, and 3- years in the training set by using “rms” and “survival” packages in software R (Figure 10A). In Figure 10B, the C-index of the nomogram of the training set was 0.650, and the AUC values predicted for 1 -, 2- and 3-year survival is 0.806, 0.844 and 0.792, respectively. The C-index was 0.615 in the testing set and the AUCs of ROC for 1-, 2-, and 3-year survival predictions were 0.653, 0.776, and 0.856, respectively (Figure10C). Likewise, the C-index was 0.618 in the whole TCGA set and the 1-, 2-, and 3-year AUCs were 0.724, 0.814, and 0.83, respectively (Figure10D). The calibration plots in (Supplementary Figure 1) exhibited excellent accordance between the nomogram prediction and the actual values in terms of the 1-, 2- and 3-year survival rates in the three datasets. The above results indicated that the prediction performance of the established nomogram is improved.
Correlation of Risk Score with TIME (Tumor Immune Environment Characterization)
Through the ESTIMATE evaluation method, TumorPurity, ImmuneScore and StromalScore were calculated. These results indicated that patients in the low-risk group have lower TumorPurity and higher ImmuneScore and StromalScore(Figure 11A,B,C). To further uncover the the correlation between GILncSig and immune cell infiltration, the analysis showed that patients in low-risk group had more T cells CD8, B cells and T cells CD4 memory activated, while the Macrophages M0 was at low level(Figure 11 D). To further explore the influence of GILncSig upon TIME of pancreatic cancer we analyzed correlation of risk signature with immune cell infiltration type and level. The results indicated that the risk signature significantly correlated with infiltrating B cells (r = -0.38; p = 1.2e − 05), infiltrating CD4+T cells (r = -0.34; p = 0.00011), infiltrating plasma cells (r = -0.19; p = 0.036), CD8 T cells (r = -0.35; p = 6.6e −05), macrophages M0(r = 0.32; p =0.00023), and macrophages M2 (r = 0.21; p = 0.018; Figure 11 E). Then, the ssGSEA algorithm was used to examine whether there was distinction of immune signatures between groups low/high risk. From the results, found that the infiltrating levels of B cells, CD8+T cells, DCs, Neutrophils, pDCs, Tfh, Th1 cells, Th2 cells were remarkably elevated and some immune signatures (i.e. CCR, checkpoint, inflammation promoting, IFN response type II) were significantly activated in the low-risk group Figure 12 A,B).
Correlation of Risk Score with Immune Checkpoint Blockade Key Molecules
Six key immune checkpoint inhibitor genes (PDCD1, CD274, PDCD1LG2, CTLA‐4, HAVCR2, and IDO1) were singled out for further research. We performed the correlation analysis of ICB key gene expression with risk signature to investigate the potential role of signature in the ICB therapy of pancreatic cancer (Figure 12 C). Correlation analysis results indicated that GILncSig had close relationship with CD274 (r = -0.18; p = 0.018), CTLA4 (r = -0.32; p = 2e −05), HAVCR2 (r = -0.29; p = 0.00012), PDCD1 (r = -0.4; p = 8.1e − 08), and PDCD1LG2 (r = -0.3; p = 6e−05; Figures 12D), indicating GILncSig might exert a nonnegligible player in ICB treatment outcome prediction in pancreatic cancer. Further correlation analysis presented that 32 of 47 (i.e. CD27, IDO2, etc.) immune check blockade–related genes expression levels were significantly different between two risk groups (Figures 13).
{kind=link}