3.1. Materials
All cell culture materials were obtained from Gibco (USA) unless otherwise specified.
3.2. Primary mouse embryonic fibroblasts (MEFs) isolation
Primary MEFs were isolated by a mechanical technique and characterized as described by our previous study (19). In short, embryonic fibroblasts with the passage number 1–5 were commonly proliferated in a culture flask pre-coated with 0.2% gelatin (Sigma, Germany) containing 10 ml of Dulbecco’s minimum eagle’s medium (DMEM; Gibco, USA) and 10% fetal bovine serum (FBS). Before stem cell culture, MEF were treated with 10 µg/ml Mitomycin C (Abcam, UK) for 3 hours to inhibit cell proliferation.
3.3. Stem cell culture and expansion
The established normal R1-hiPSC and hESC XX Royan H1 cell lines (20) were used in this study. The pluripotent stem cells (PSCs) culture media was composed of 75% DMEM/F12 supplemented with 20% knockout serum replacement, 1% non-essential amino acids, 2 mM L-glutamine (Sigma, Germany), 0.1% β-mercaptoethanol (Merk, Germany), 1% insulin/transferrin/selenium solution, and 12 ng/ml basic human recombinant FGF (b-FGF) (Royan, Iran). The standard cultures were split at the ratio of 1:5 every 7–10 days.
3.4. Endoderm differentiation
Upon 70–80 confluence, hiPSCs were detached using 1 mg/ml Collagenase Type IV (Stem Cell Technologies, Canada) and dissociated into single cells by gentle pipetting. The cells were washed with phosphate-buffered saline (PBS) and suspended in fresh ESCs culture media containing 100 ng/ml of b-FGF, and then transferred on Matrigel- (Sigma, Germany) coated 60 mm plates. The next day, the medium was discarded and the differentiation medium consisted of RPMI-1640 with 100 ng/ml activin A (BioLegend, USA) with varying concentrations of defined-FBS (D-FBS) used. The concentrations of D-FBS were set to 0% (Day 0), 0.2% (Day 1), and 2% (Day 2 and 3).. The cells were harvested and used for different analyses at the fourth 24h (Day 4) (21).
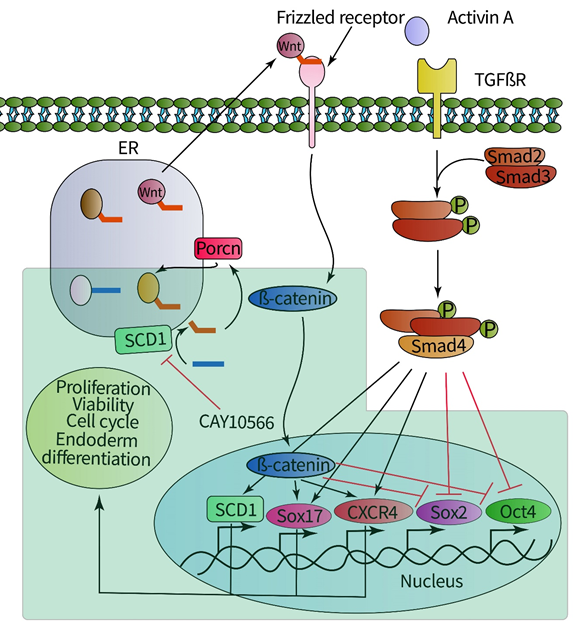
3.5. Targeting SCD1 by small molecule inhibitor
Chemical inhibition of SCD1 was performed with the specific inhibitor of SCD1 CAY10566 (Cayman Chemical, USA). Cells were treated with SCD1 inhibitor for 24 hours at days 0, 1, or 2 of the differentiation procedure. In our previous study, the possible toxicity of the inhibitor was determined using Trypan Blue exclusion and sulforhodamine B assays after treatment with 25 nmol/L CAY10566 or lower concentrations (17). The rescue experiments were performed using 50 µM oleate.
3.6. Evaluation of cell viability
An Annexin V-FITC/PI apoptosis detection kit (eBioscience, USA) was used for the apoptosis analysis. After completion of the endodermal differentiation procedure, cells were detached with collagenase IV and washed once with PBS. The cells were suspended in the binding buffer (1X), incubated with 5 µl Annexin-V for 10 minutes, and washed once with binding buffer. The cells were re-suspended again in 200 µl of binding buffer followed by the addition of 10 µl of propidium iodide. The percentage of apoptosis and necrosis was determined using a flow cytometer (Miltenyi Biotec, USA) and results were analyzed using flow Jo 7.6.1 software (Tree Star, USA).
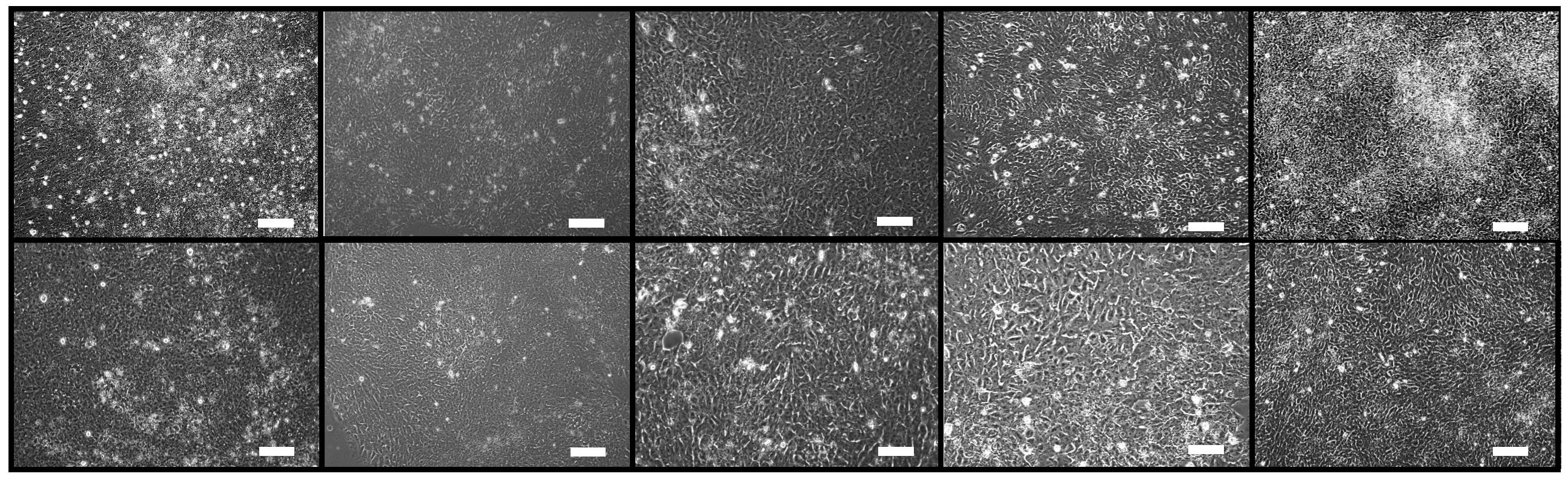
3.7. Morphological examination
The morphological alteration was visually monitored using a Cytation 5 Cell Imaging Multi-Mode Reader system (BioTek, USA). Differentiating cells were imaged every 24 hours for consecutive 4 days in the absence or the presence of CAY10566.
3.8. Gene expression assay
Total RNA extraction was carried out using an RNA extraction kit (TaKaRa, Japan) following the instructions provided by the manufacturer. The quantity and quality of extracted RNA were checked using a spectrophotometer (NanoDrop Technologies, USA) and running on 1.5% agarose gel, respectively. The complementary DNA (cDNA) required for real-time PCR was synthesized with a cDNA synthesis kit (Roche, UK). Real-time PCR was performed with SYBR Green PCR master mix (Yekta Tajhiz Azma, Iran) on a MIC real-time PCR system (BioMolecular Systems, Australia). The primer sequences used for Sox2, Oct4, Sox17, and CXCR4 expression assay are listed in Supplementary Table 1. Relative gene expression was normalized against the expression level of GAPDH as a reference gene for each sample and all alterations were expressed as fold-changes relative to the mock.
3.9. Surface markers analysis
The pellet of disassociated cells was resuspended in 4% paraformaldehyde to fix the cells by a 30 minutes incubation at 4°C. Cells were then resuspended in PBS containing 1% BSA as a staining buffer and incubated for 20 minutes at 4°C. Then, cells were incubated with Alexa Fluor 488-conjugated CXCR4 (R&D, USA) and PE-conjugated KDR (R&D, USA), Alexa Fluor 488-conjugated SSEA-3, (eBioscience, USA), or PE-conjugated NCAM antibodies at 4°C for 1 hour. After PBS washes, cells were analyzed using a flow cytometer (Miltenyi Biotec, USA) and the data were analyzed by the Flowing Software 2.5.1 (Turku Bioscience, Finland).
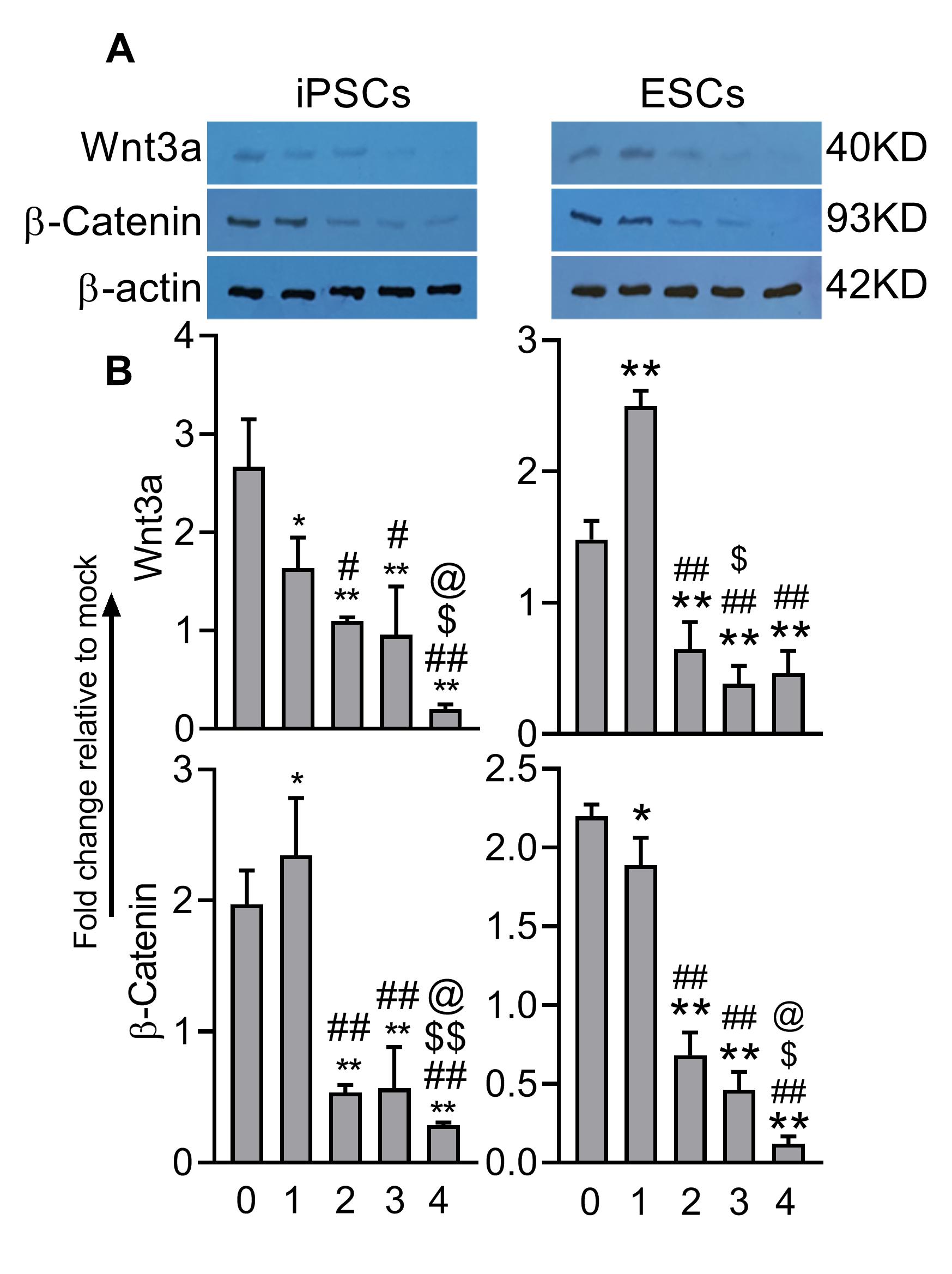
3.10. Western blotting
The cell lysate from each group was prepared using a RIPA lysis buffer containing protease inhibitor. Samples were centrifuged and the total protein concentration of supernatants determined by Lowry protein assay. Twenty µg of protein was mixed with an equal volume of sample buffer and electrophoresed on SDS-PAGE, then transferred onto PVDF membranes (Santa Cruz, USA). After blocking with 5% skimmed milk, the membrane was washed in PBS with Tween detergent and treated with primary antibodies against the pluripotency markers Oct4 (Abcam, USA) and Sox2 (Abcam, UK), endodermic markers CXCR4 (Santa Cruz, USA) and Sox17 (Santa Cruz, USA), and the internal control β-actin (Santa Cruz Biotechnology, USA) overnight at 4°C. Primary antibodies against Wnt3a (Santa Cruz, USA) and β-catenin (Santa Cruz) were applied for evaluation of Wnt signaling pathway. Then, the membrane was exposed to horseradish peroxidase (HRP)-conjugated secondary antibody for 1 hour at RT. Finally, the membrane was visualized by luminol reagents (Santa Cruz, USA). The intensity of the bands was quantified using ImageJ software (version 1.41).
3.11 Cell proliferation assay
The proliferation rate was measured using a 5-bromo-2-deoxyuridine (BrdU) assay (Abcam, USA). To this end, cells were seeded at a density of 2×104 cells per well of 96-well plates. After completion of the treatment protocol, each well was filled with BrdU reagent before cell harvesting. Cellular DNA was denatured with the fixing solution at RT. After washing with PBS, the anti-BrdU monoclonal antibody was added to each well, and plates were incubated for 1 hour at RT. Then, supernatants were discarded and the washing step was repeated followed by the addition of an HRP-conjugated anti-IgG antibody. After the final wash, each well was incubated with a peroxidase substrate and then the reaction was stopped by adding the stop solution when the yellow color was generated. The optical density of the solution was measured using a microplate reader (Labsystems, Finland) at 450 nm. Wells containing media alone and seeded cells without BrdU reagent were used as blank and background controls, respectively.
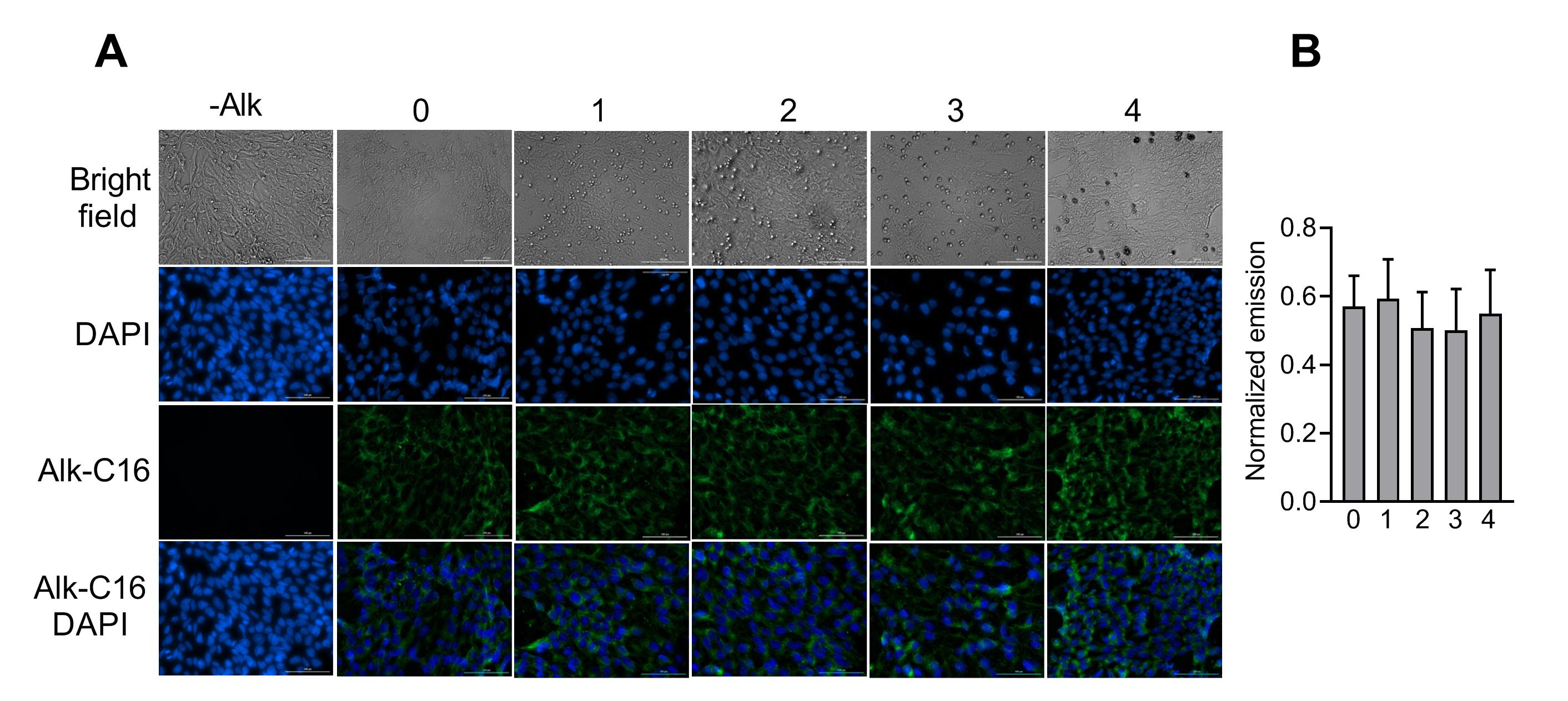
3.12 Acylation assay
In order to evaluate the total protein acylation, we performed an Alkyne-azide cycloaddition click reaction according to a standard protocol (22). Briefly, cells were seeded onto a 24-well plate at a density of 5×104 cell/well and treated as abovementioned. Medium containing the ω-alkynyl analog of palmitic acid (Alk-C16) was added into the wells at each time point after washing once with PBS and incubated for 24 hours at 37°C with 5% CO2 to label the cells. After discarding the medium, cells were then washed with pre-cooled PBS, fixed with − 20°C pre-chilled methanol. Permeabilization was performed using Triton X-100 at RT for 5 minutes. Cells were washed with PBS and exposed to a click labeling reagent containing Alexa Fluor 488 Azide (Invitrogen, USA), Tris (2-carboxyethyl) phosphine hydrochloride (TCEP) (Cayman, USA), and CuSO4 for 1 hour at RT and dark. The wells were then washed with PBS, incubated with 4′,6-diamidino-2-phenylindole (DAPI) for 30 S at RT, and washed three times with PBS. Finally, cells were imaged using the cell imaging system at excitation 488/emission 516 nm for Alexa Fluor488 and excitation 377/emission 477 nm for DAPI.
3.13 Cell cycle analysis
Cell cycle assay was performed using a cell cycle phase determination kit (Cayman Chemical, USA). Cells were seeded on the culture plates according to the manufacturers' instructions. After completion of treatment protocols, the cells were trypsinized, centrifuged, and washed with the assay buffer. The cell pellet was resuspended in the assay buffer and incubated with a fixation buffer. Then, the cell suspension was centrifuged and the supernatant was discarded followed by incubation in staining solution at RT and dark. The percentage of cells in each phase was determined using Flowing software 2.5.1 (Turku Centre for Biotechnology, Finland).
3.14 Statistical Analysis
Experimental data are presented as mean ± standard deviation (SD) of the mean from at least three independent experiments. One-way or two-way analysis of variance (ANOVA) followed by Turkey’s post-hoc test was applied for Groupwise comparisons (GraphPad Software 8.0, USA). A p-value less than 0.05 was considered to be significant.
{kind=link}
{kind=link}
{kind=link}
{kind=link}