Distinctive lncRNA and mRNA expression between normal ovary and OSC tissues
The lncRNA and mRNA expression patterns were compared between normal ovary tissues and OSC samples. Normal ovary tissues (n = 8) and OSC samples (n = 8) were subjected to the RVM t-test. A total of 2,939 lncRNAs and 2,766 mRNAs with significant differential expression between OSC and normal ovary tissues (fold change ≥ 2.0 or ≤ 0.6, P < 0.05; Supplementary Tables 2 & 3) were identified. Of these 2,939 lncRNAs, 1,655 were downregulated and 1,284 were upregulated. The differences indicated their potential roles in OSC pathogenesis. Hierarchical cluster analysis showed systematic variations in the expression of differential lncRNAs and mRNAs in OSC and normal ovary tissues (Fig. 1a). Thus, the lncRNA and mRNA expression signatures were likely representative.
Construction of a lncRNA-mRNA co-expression network and prediction of lncRNA functional roles based on this network
A co-expression network was built to analyze the interactions between lncRNAs and mRNAs. The workflow of the construction of the lncRNA-mRNA co-expression network is shown in Fig. 1b. First, the main functions of the 2,766 differentially expressed mRNAs in OSC were determined by GO analysis. The results showed that 2,090 mRNAs were involved in 362 upregulated and 160 downregulated significant functions (P < 0.05; Supplementary Table 4). The pathways of the altered 2766 mRNAs involved were then analyzed according to KEGG. A total of 589 mRNAs were involved in the 59 enrichment-related pathways (P < 0.05; Supplementary Table 5), 45 of which were upregulated and 14 were downregulated.
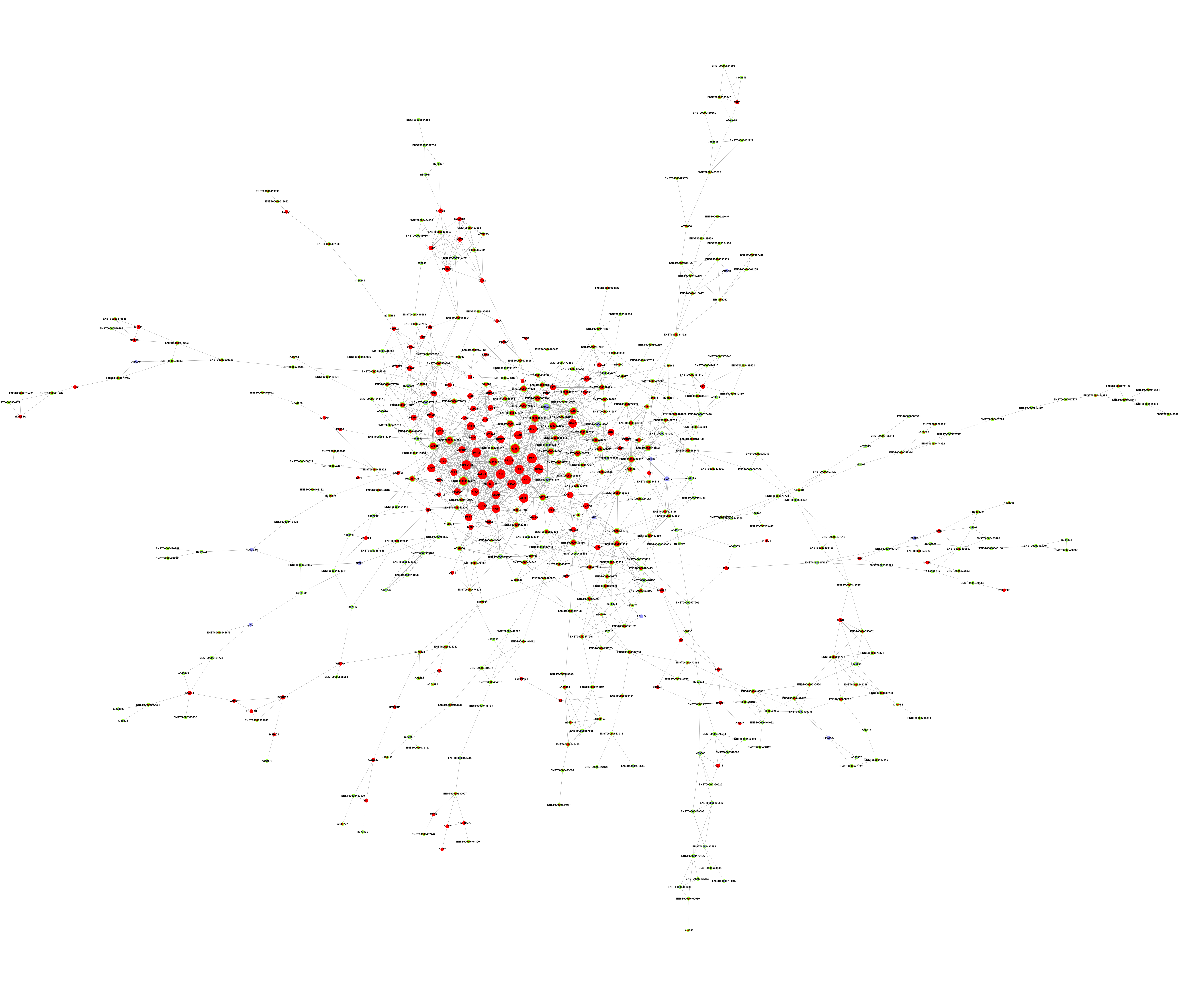
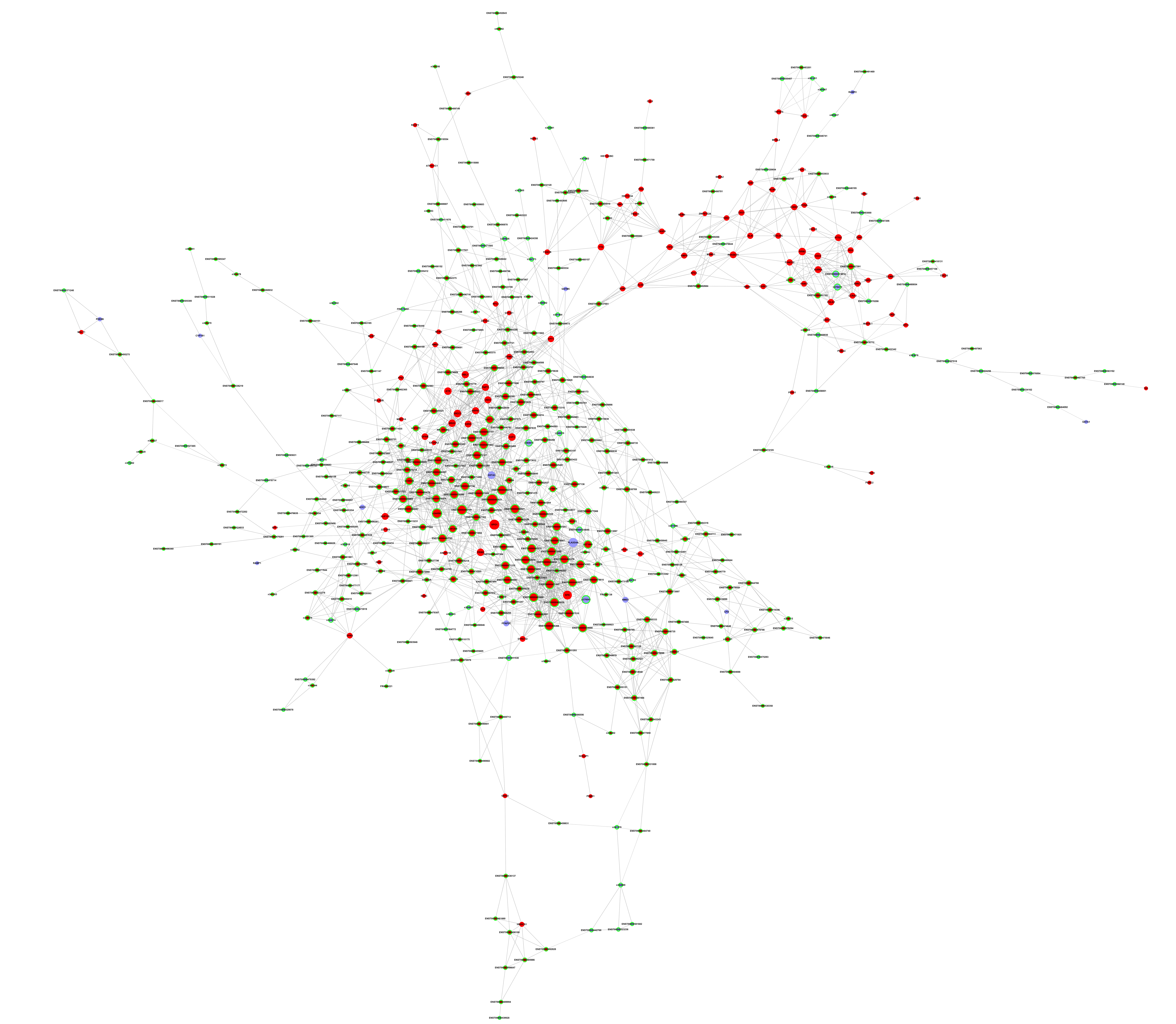
As a result, 229 mRNAs (Supplementary Table 6) with significant functions and pathways in GO and pathway analysis were termed GO&Pathway mRNAs. The levels of the GO&Pathway mRNAs and the differentially expressed lncRNAs in the normal ovary and OSC groups were used to construct two lncRNA-mRNA co-expression networks (Supplementary Figs. 1 & 2). The mRNAs and lncRNAs involved in the two networks were sorted by degree and clustering coefficient as shown in Supplementary Tables 7 & 8.
To identify lncRNAs involved in OSC, the two lncRNA-mRNA co-expression networks were compared, and the difference in relative degree (∣DiffK∣) was calculated (Supplementary Table 9). The ∣DiffK∣ represents the change in the common expression status of lncRNAs in two networks. As listed in Supplementary Table 10, 67 lncRNAs (with a ∣DiffK∣ >0.25) were selected for further functional analysis.
According to the lncRNA-mRNA co-expression networks in the normal ovary and OSC groups, 109 mRNAs were co-expressed with the selected 67 lncRNAs. KEGG pathway analysis showed that the 109 mRNAs were involved in 32 upregulated and 4 downregulated pathways involved in various biological processes, including cellular processes, environmental information processing, human diseases, genetic information processing, metabolism, and immune system (P < 0.05; Fig. 2).
Based on these pathways, we predicted the potential roles of the 67 lncRNAs in OSC pathogenesis. Co-expressed lncRNAs and mRNAs in upregulated pathways were shown in the phagosome, focal adhesion, apoptosis, cell cycle, regulation of actin cytoskeleton, progesterone-mediated oocyte maturation, ECM-receptor interaction, proteoglycans in cancer, pathways in cancer, PI3K-Akt, VEGF, p53, and oocyte meiosis signaling pathways (Fig. 3a, b; Supplementary Table 11); in the DNA replication, viral carcinogenesis, homologous recombination, mismatch repair, ribosome biogenesis in eukaryotes, RNA transport, nucleotide excision repair, proteasome, HIF-1, Fanconi anemia, and ubiquitin-mediated proteolysis signaling pathways (Fig. 3c, Supplementary Table 12); in thecytokine-cytokine receptor interaction, progesterone-mediated oocyte maturation, B cell receptor, chemokine, and Toll-like receptor signaling pathway (Fig. 3d, Supplementary Table 13); and in the purine metabolism, pyrimidine metabolism, tryptophan metabolism, biosynthesis of amino acids, and Metabolism pathways (Fig. 3e, Supplementary Table 14). Detailed descriptions of 28 mRNAs which participated in multiple pathways in the co-expression network are provided in Supplementary Table 15. The PIK3CA, KRAS, and NRAS genes play key roles in OSC progression. KRAS and NRAS function in seven signaling pathways; PIK3CA is involved in eleven signaling pathways, seven of which are common with KRAS and NRAS pathways. Co-expressed lncRNAs and mRNAs in downregulated pathways were involved in drug metabolism, lipid metabolism, tyrosine metabolism, and ABC transporter signaling pathways (Fig. 3f, Supplementary Table 16).
Taken together, these results demonstrated that lncRNA-regulated mRNAs were linked to multiple biological processes that affected tumor cell proliferation, apoptosis, migration, invasion, and angiogenesis.
Identification of the possible roles of lncRNAs based on genomic co-localization relative to protein coding genes
To further explore the possible roles of differentially expressed lncRNAs during the development of OSC, we analyzed their genomic locations and classified them into lincRNA, antisense lncRNA, or enhancer-like lncRNA. Among the 2,939 differentially expressed lncRNAs between OSC and normal ovary tissues, 166 were lincRNAs, 144 were antisense lncRNAs, and 43 were enhancer-like lncRNAs. We combined these lincRNAs, antisense lncRNAs, and enhancer-like lncRNAs with their adjacent coding genes located within 300 kb in the genome and differentially expressed in OSC to analyze the potential functions of these lncRNAs. The lncRNAs and associated coding genes are shown in Supplementary Table 17.
LincRNAs are large intergenic non-coding RNAs (Fig. 4a). The functions of 144 lincRNAs were predicted through pathway analysis of adjacent genes. The results indicated that 23 lincRNAs may regulate the expression of 26 coding genes involved in 20 upregulated pathways (Fig. 4b). These pathways regulate cell adhesion, invasion, migration, proliferation, and apoptosis among cellular processes, genetic and environmental information processing, and metabolism (Fig. 4c-l). The coding genes SOS1 and ITGB1 may play a key role in tumor progression, and they were involved in more than six signaling pathways.
We then analyzed 144 antisense lncRNAs (Fig. 5a). KEGG pathway analysis was used to predict the biological roles of 19 antisense lncRNAs, and the results indicated that 16 coding genes were involved in 15 upregulated pathways and 2 downregulated pathways (Fig. 5b, c). These pathways are involved in cellular processes, metabolism, and environmental and genetic information processing. Of the 16 coding genes, RAD51, COL4A1, and BIRC2 were involved in more than one signaling pathway (Fig. 5d-j).
Finally, we predicted the roles of enhancer-like lncRNAs in OSC (Fig. 5K). Of 43 enhancer-like lncRNAs, four were positively correlated with the expression of five mRNAs. Therefore, the functions of four lncRNAs that acted as enhancers were predicted through pathway analysis of five coding genes. The results indicated that the coding genes BMS1 and ANAPC1 participated in four upregulated pathways (Fig. 5l). These pathways represent cellular processes and genetic information processing, including cell cycle, oocyte meiosis, ubiquitin-mediated proteolysis, and ribosome biogenesis in eukaryotes. The ubiquitin system regulates cell differentiation and immunity, and is involved in transcription, regulation of secretion, and cell development by mediating protein degradation. We predicted that two enhancer-like lncRNAs (n371668 and n378384) played important roles in OSC cell proliferation (Fig. 5m).
{kind=link}
{kind=link}