Soybean seedling growth conditions and simulated drought stress treatment
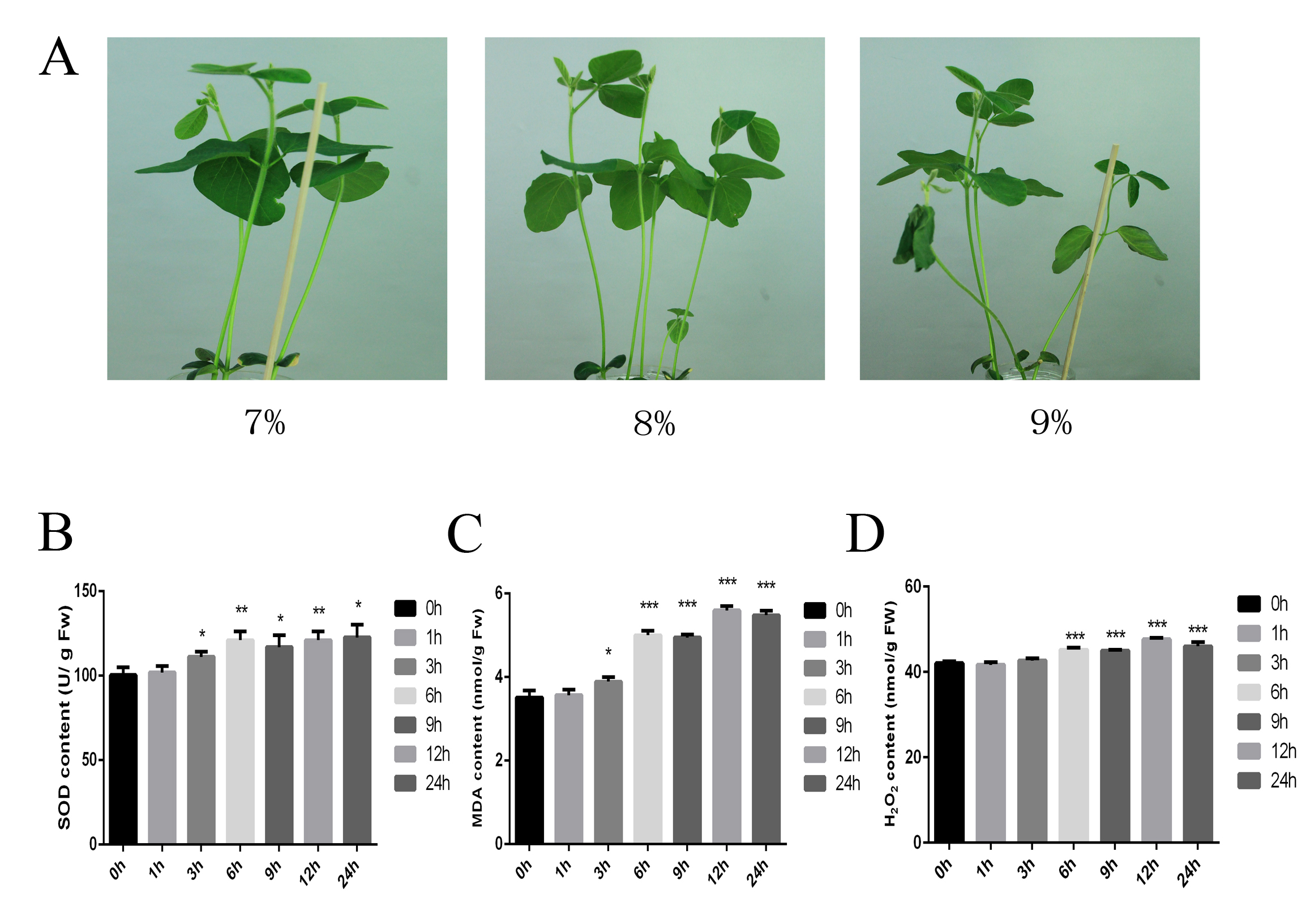
The cultivated soybean seeds of Glycine max (Williams 82) provided by Northeast Institute of Geography and Agroecology, Chinese Academy of Sciences were used for our study. The seeds were surface-sterilized in ethanol for at least 10 min and then washed several times with distilled water. An equal number of seeds were added to pots containing half-strength (1/2) Hoagland’s nutrient solution and incubated at 25°C/18°C (day/night) with a 16-h/8-h (light/dark) photoperiod under photosynthetic photon flux density of 80 μmol m−2 s−1. Once the first compound leaf had expanded, three replicates of each group were exposed to 7%, 8%, or 9% polyethylene glycol (PEG) 8000 for 1, 3, 6, 9, 12, or 24 h to simulate drought. An untreated (0 h) pot was used as a control. Physiological indices were measured using superoxide dismutase (SOD), malondialdehyde (MDA), and catalase (CAT) detection kits (A001-1, A003-1, and A007-1, Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Notice that, the phenotype showed that the leaves of seedlings treated with 9% PEG 8000 were wilted substantially, and the SOD activity and contents of MDA and H2O2 were significantly affected at 6 and 12 h (Figure S6). Therefore, all samples of three replicates treated with 8% PEG 8000 at 0, 6, and 12 h were used to prepare RNA libraries.
Total RNA extraction and mRNA, degradome and sRNA library preparation
The RNA sequencing (RNA-seq) experiment was conducted using three replicates. Leaf and root samples were collected from seedlings treated with 8% PEG 8000 for different durations. All samples were immediately frozen in liquid nitrogen and stored at −80°C until analyzed. Total RNA was extracted using the TRIzol® Reagent (Invitrogen, Carlsbad, CA, USA). The concentration, purity, and integrity of the total RNA were analyzed using a NanoPhotometer® spectrophotometer (Implen, Westlake Village, CA, USA), the Qubit™ RNA Assay Kit with a Qubit 2.0 Fluorometer (Life Technologies, Camarillo, CA, USA), and the RNA Nano 6000 Assay Kit with the Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA).
Transcriptome library construction and sequencing
The mRNA-seq experiment was completed using three replicates of 18 samples for the leaf and root samples collected at 0, 6, and 12 h. The mRNA-seq libraries were prepared from 1 μg RNA per sample. The libraries were sequenced using the HiSeq X™ Ten platform. The paired-end reads were filtered by removing the adapters and low-quality sequences. The remaining high-quality reads were mapped to the Glycine_max.Wm82.a2.v1 reference genome (http://phytozome.jgi.doe.gov/pz/portal.html). The mapped reads were further analyzed using the TopHat2 software. First, the mapped reads were spliced using the Cufflinks program and new genes were detected by filtering out sequences that encoded short peptide chains (fewer than 50 amino acids) or contained only one exon. Furthermore, the sequences were compared with those of the non-redundant (nr), Swiss-Prot, Clusters of Orthologous Groups of proteins (COG), Clusters of Protein homology (KOG), and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases using the BLASTX online tool. The GO annotations of genes assigned by the Blast2GO software were based on the results of the comparison with nr database protein sequences. KEGG Orthology information for the genes was obtained using KOBAS 2.0 [42]. The amino acid sequences encoded by the genes were compared with protein sequences in the Pfam database using the HMMER program for the annotations [43]. The mRNA levels were normalized based on the fragments per kilobase of transcript per million mapped reads (FPKM) value. The differentially expressed genes (DEGs) were detected based on the following parameters: |log2FC| ≥ 1 with FDR < 0.05 and CPM > 1.
Small RNA sequencing and miRNA identification
For the sRNA-seq experiment, mRNA libraries were prepared for the leaf and root tissues collected at 0, 6, and 12 h. Purified RNA (1.5 μg) was used to construct a sequencing library with the NEBNext® Ultra Small RNA Sample Library Prep Kit for Illumina® (New England Biolabs, Ipswich, MA, USA). The libraries were sequenced using the Illumina HiSeq 2500 platform. The resulting paired-end reads were filtered by removing contaminants and low-quality reads, and reads containing poly-Ns and adapters. In addition, sequences shorter than 18 nucleotides or longer than 30 nucleotides were eliminated to ensure the accuracy of all downstream analyses. Using the Bowtie software, the clean reads were mapped based on sequence alignments using the SILVA, GtRNAdb, Rfam, and Repbase databases to filter out ribosomal RNA, transfer RNA, small nuclear RNA, small nucleolar RNA, and other non-coding RNA and repeats. The remaining reads were used to detect known miRNAs based on comparisons with miRNAs in the miRBase database (version 21.0) (http://www.mirbase.org/). The miRDeep-p tool was applied to predict plant novel miRNAs [44]. Potential precursor sequences were obtained by analysing the genomic position information. Based on the distribution of the miRNA characteristics (i.e., mature, star, and loop regions) as well as precursor energy information obtained with the RNAfold and randfold tools [45], we scored the newly identified miRNAs using a Bayesian model. The frequency of the miRNAs in all libraries was expressed as transcripts per million (TPM). The differentially expressed miRNAs (DEMs) were detected based on the following parameters: |log2FC (fold change)| ≥ 1 with false discovery rate (FDR) < 0.05 and counts per million (CPM) > 1.
Degradome sequencing, target identification, and data analysis
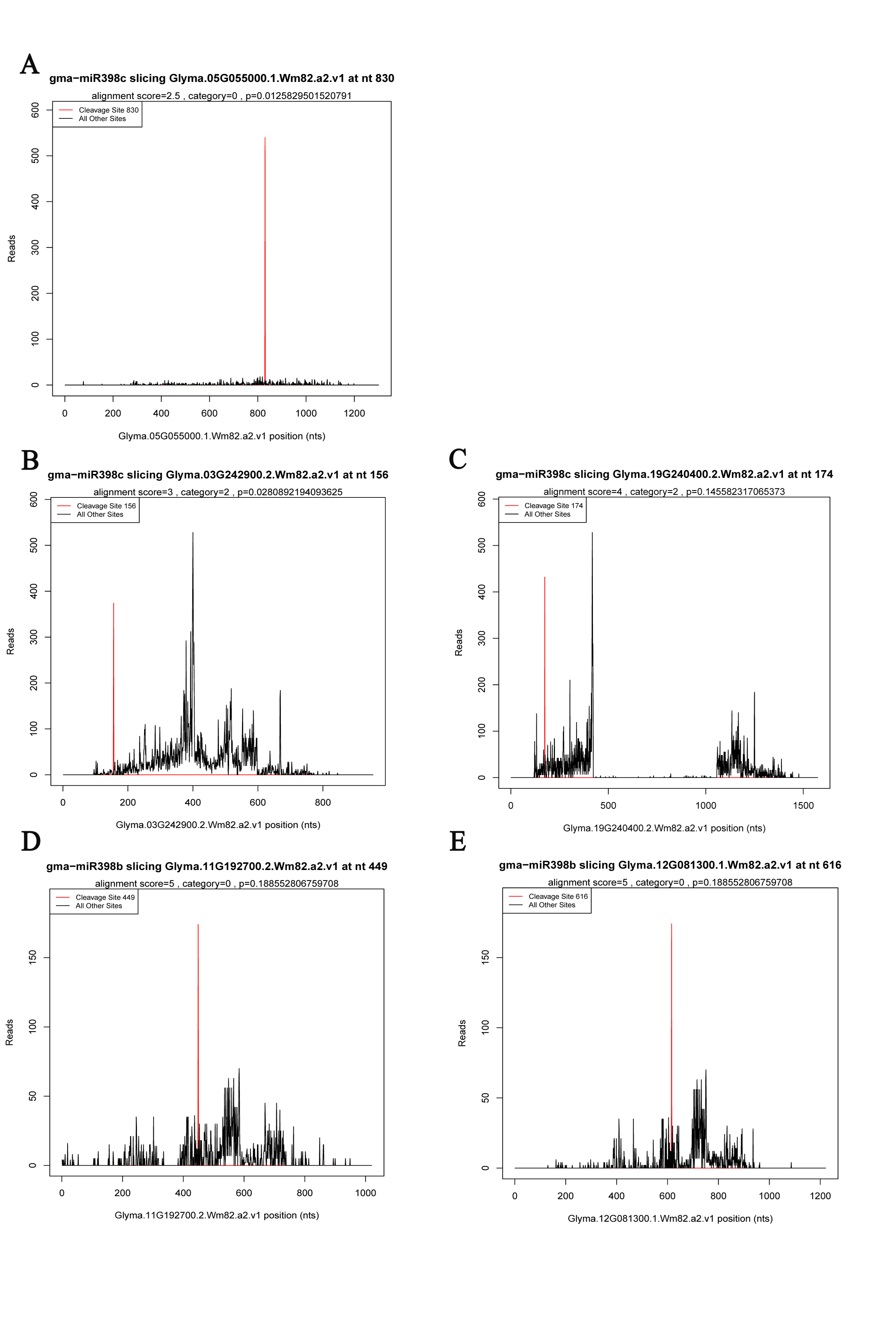
The leaf and root samples collected at different time points were respectively mixed to obtain two pools for degradome sequencing. The degradome libraries were prepared using approximately 25 μg total RNA from the mixed leaf and mixed root samples [46]. Biotinylated random primers were annealed to poly-A+ RNA, which was used as the template. The RNA fragments with the biotin tag were obtained using streptavidin. Only RNAs with 5′ monophosphates were ligated with a 5′ adapter. The resulting RNA fragments were used as templates for first-strand cDNA synthesis. In addition, sequencing primers were designed consistent with the 5′ adapter sequence, which corresponded to the first 50 nucleotides from the 5′ site of the cleaved RNAs. Sequencing (50 bp single-end reads) was performed using the HiSeq 2500 platform. The spliced miRNA targets were identified and classified using the CleaveLand 3.0 pipeline. Putatively identified transcripts were divided into five categories: category 0: more than one raw read at the cleaved site, abundance at the site is equal to the only maximum on the transcript; category 1: more than one raw read at the cleaved site, abundance at the site is equal to a maximum on the transcript and there is more than one maximum; category 2: more than one raw read at the cleaved site, abundance at the site is less than the maximum but more than the median value for the transcript; category 3: more than one raw read at the cleaved site, abundance at the site is less than or equal to the median value for the transcript; and category 4: only one raw read at the cleaved site [47]. The potential target genes were subjected to gene ontology (GO) analysis using the AgriGO toolkit [48].
Verification by RT-qPCR analysis
The total RNA extracted from plants treated with 8% PEG 8000 (0, 6, and 12 h) was used for quantitative reverse transcription PCR (RT-qPCR) and stem-loop RT-qPCR analyses of genes (mRNA) and miRNAs, respectively, in accordance with the MIQE standards [49]. All primer sequences were designed and synthesized based on the miRNA and mRNA sequences (Table S7) (Genewiz, Beijing, China). For the RT-qPCR analysis of genes, total RNA samples were treated with RNase-free DNase I to eliminate any contaminating genomic DNA. The RNA was then used as the template for cDNA synthesis with the PrimeScript™ RT Reagent Kit with gDNA Eraser (Takara). The stem-loop RT-qPCR analysis of miRNA involved a special step during which 1 µg RNA was reverse-transcribed with PrimeScript RT Enzyme Mix I and miRNA-specific stem-loop primers. The RT-qPCR and stem-loop RT-qPCR were completed using the SYBR® Premix Ex Taq™ II kit (Takara). We selected GmActin and GmEF1b for RT-qPCR, and gma-miR1520d for stem-loop RT-qPCR as internal controls. The RT-qPCR and stem-loop RT-qPCR experiments were completed with three technical replicates for each biological replicate. Relative expression levels were quantified using the 2−ΔΔCt method.
Analysis of the interaction between gma-miR398 and their targets
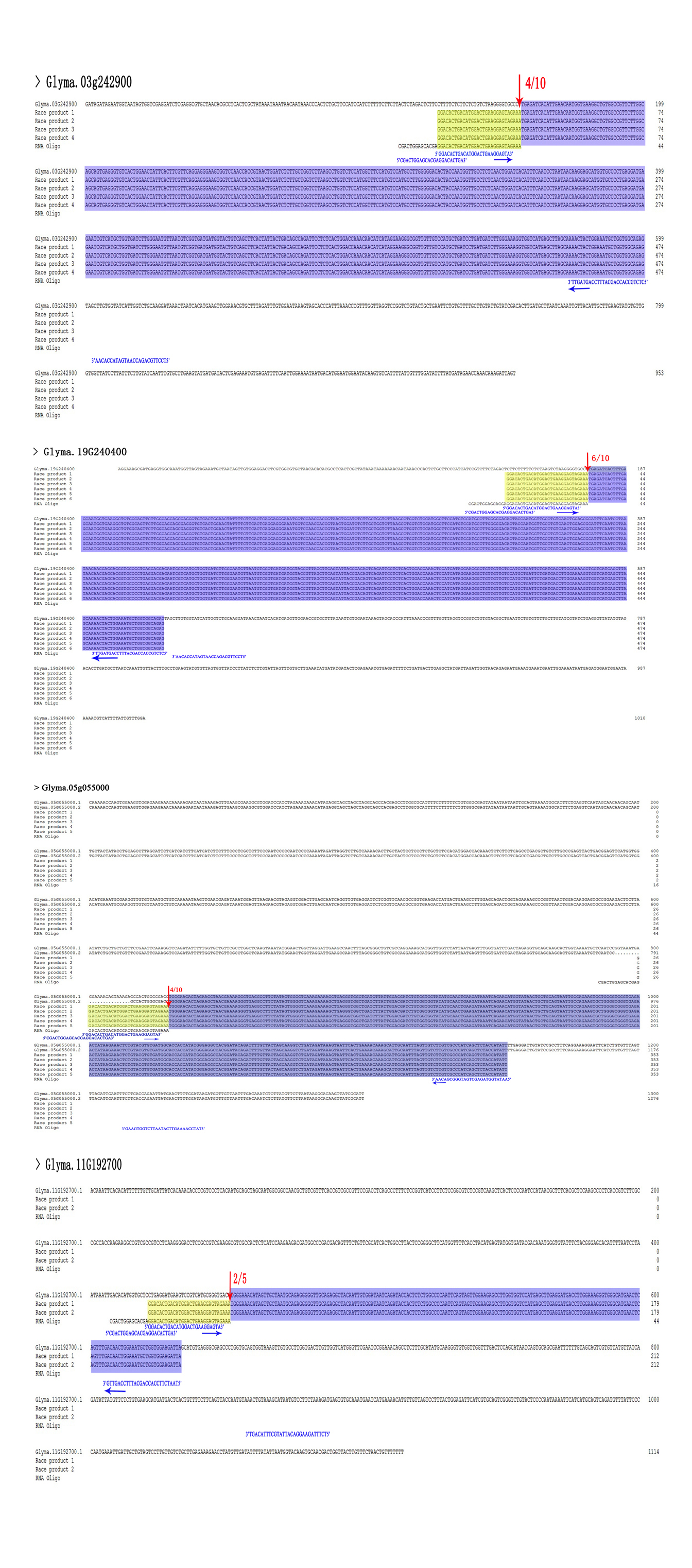
A modified RNA ligase-mediated 5′ rapid amplification of cDNA ends (RACE) completed with the GeneRacer™ Kit (Invitrogen) was used to examine RNA cleavage. First, the RNA oligo was ligated to 3 µg total RNA from each mixed leaf and mixed root sample. Using random primers, we generated cDNA from the mRNA with the 5′ RACE adapter by reverse transcription. The GSP and GSP-Nested primers from the kit were used to amplify the 5′ end of the cleaved transcript with two gene-specific primers by nested PCR and touchdown PCR. Finally, the nested PCR product was incorporated into the pEASY-T1 vector (Table S7) (TransGen, Beijing, China), inserted into Escherichia coli (DH5α) cells and sequenced.
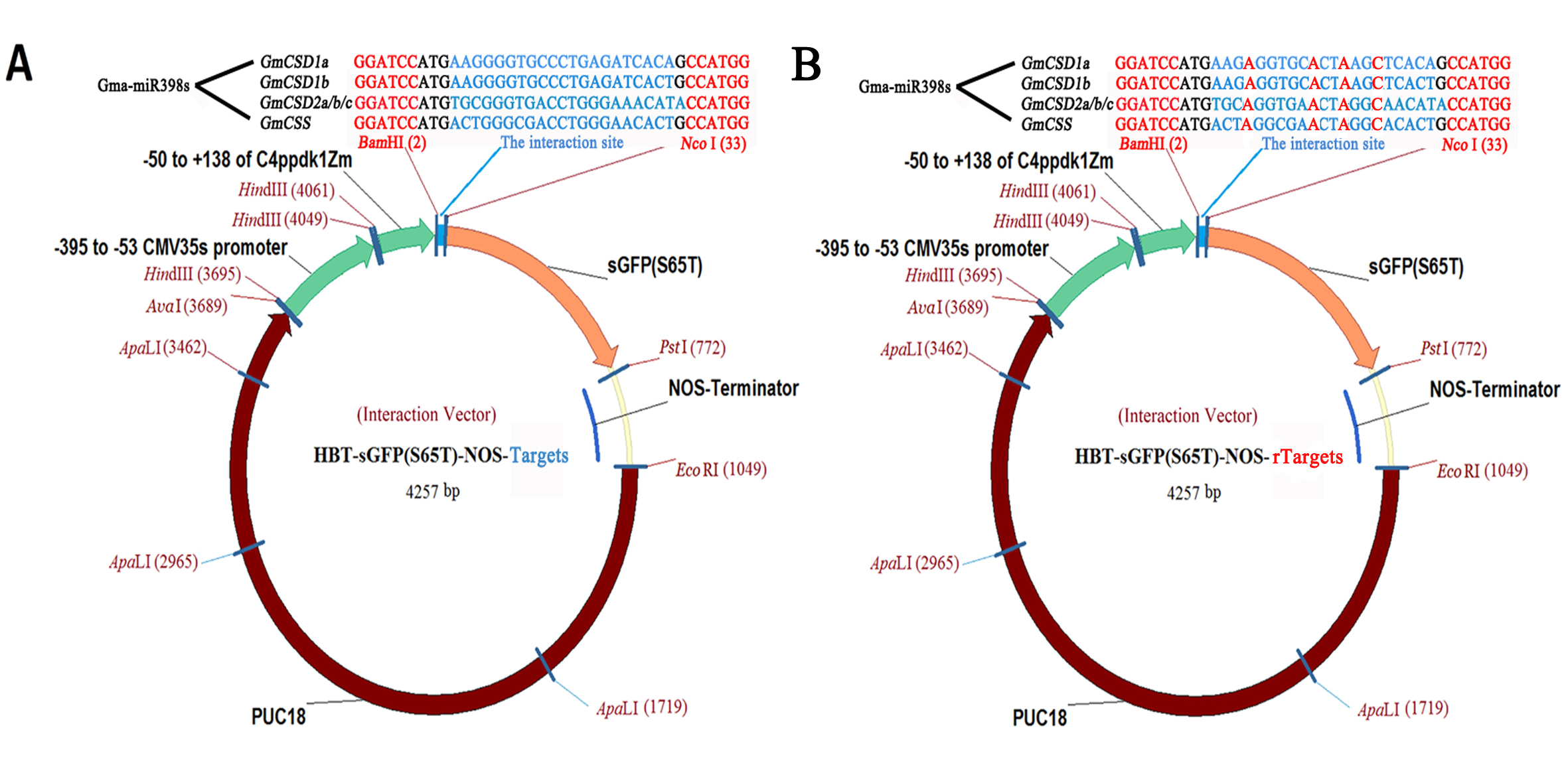
The Wild-type Arabidopsis thaliana (ecotype Columbia) from Northeast Institute of Geography and Agroecology, Chinese Academy of Sciences was used for all Arabidopsis experiments. Arabidopsis mesophyll protoplasts were transiently transformed to clarify the interactions between miR398 and their targets [6]. We synthesized four interaction sequences (i.e., for GmCSD1a, GmCSD1b, GmCSD2a/b/c, and GmCCS) and four synonymous mutation sequences and inserted them into the HBT-sGFP(S65T)-NOS vector (Figure S7). Mesophyll protoplasts were extracted from 4-week-old Arabidopsis, wild-type Arabidopsis (WT), empty vector overexpression plants (OE-vector), and gma-miR398c overexpression plants (OE-miR398c). Following DNA-PEG-calcium–mediated transformation [50], the green fluorescent protein (GFP) and chloroplast autofluorescence signals were observed using an IX51 inverted fluorescence and phase contrast microscope (Olympus, Japan).
Subcellular localization of GmCSDs and GmCCS
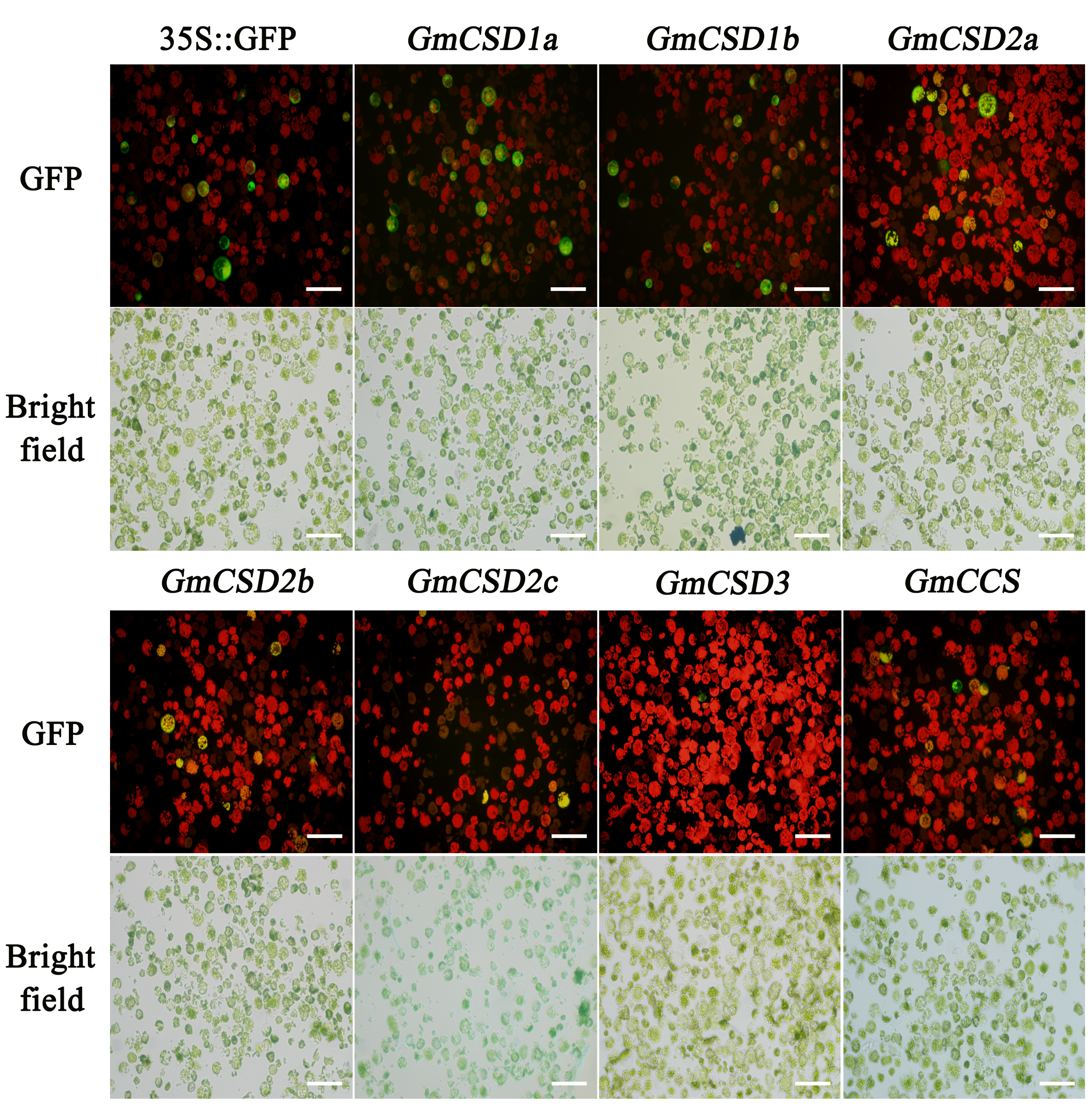
Firstly, we constructed a phylogenetic tree consisting of GmSOD-related genes to characterize potential members, and 14 distinct open reading frames potentially encoding 13 GmSOD genes and one GmCCS gene were detected in soybean. These genes contribute to the degradation of reactive oxygen species (ROS) (Figure S5). To determine the subcellular localization of GmCSDs and GmCCS via transient expression in tobacco leaves and Arabidopsis mesophyll protoplasts, the coding regions were cloned into the pCAMBIA-1302 vector and HBT-sGFP(S65T)-NOS vector for fusion with GFP, respectively (Table S7). The Agrobacterium tumefaciens strain EH105 harboring 35S:GmCSDs-GFP, 35S:GmCCS-GFP, or 35S:GFP was separately transformed into tobacco leaves, and a nucleus marker (NLS) [51] and peroxisome marker (SKL) [52] were used to co-localize with functional genes. After 48 h, leaf epidermal cells from transformed tobacco were observed with a confocal microscope (C2-ER, Nikon, Japan). Fluorescence of GFP, chloroplasts, and the markers was stimulated at 488, 640, and 561 nm, respectively.
Generation of transgenic Arabidopsis thaliana and simulated drought stress treatment
The gma-miR398c precursor sequence (gma-MIR398c) was inserted in the pCAMBIA-3301 vector using the BglII and PmlI sites between the CaMV-35S promoter and Nos-terminator. Wild-type Arabidopsis was then transformed with either the empty vector or the vector carrying gma-MIR398c using the EHA105 strain Agrobacterium tumefaciens floral-dip method. Seeds from T2 transgenic lines exhibiting a segregation ratio of 3:1 and from three homozygous T3 lines with the highest transgene expression levels were screened on Murashige and Skoog (MS) medium supplemented with BASTA (10 mg l−1 active ingredient).
Arabidopsis seeds were surface-sterilized and imbibed at 4°C for 48 h in darkness. The seeds were then sown on MS medium supplemented with 3% sucrose (w/v), 0.8% agar (w/v), and 250 mM or 300 mM D-mannitol to induce low-water-potential stress. The germination frequency (%) was calculated after treatment for 14 days, and survival rate of Arabidopsis seedlings after 10 days cultivation on 300 mM D-mannitol medium. The plates were incubated at 22°C under a 16-h/8-h (light/dark) photoperiod. For simulated drought treatment of soil-grown plants, an equal number (25:25) of transgenic and WT Arabidopsis plants were cultivated side-by-side in the same container containing soil:vermiculite (7:3) in a greenhouse at 22°C under a 12-h/12-h (light/dark) photoperiod with 50% relative humidity. Importantly, water was withheld from 3-week-old Arabidopsis plants until they wilted (i.e., for at least 3 weeks). The watering of seedlings was resumed and plants were analyzed after 3 days. Water loss was measured using detached leaves collected from 4-week-old Arabidopsis plants. The detached leaves were weighed at 1-h intervals at room temperature (22°C) under normal light with 70% relative humidity. The water loss rate was calculated based on the percentage of the initial fresh weight that remained at different time points. The germination, survival, and water loss rates for five independent replicates of each transgenic line were estimated.
Agrobacterium Rhizogenes-mediated soybean hairy roots transformation and drought stress
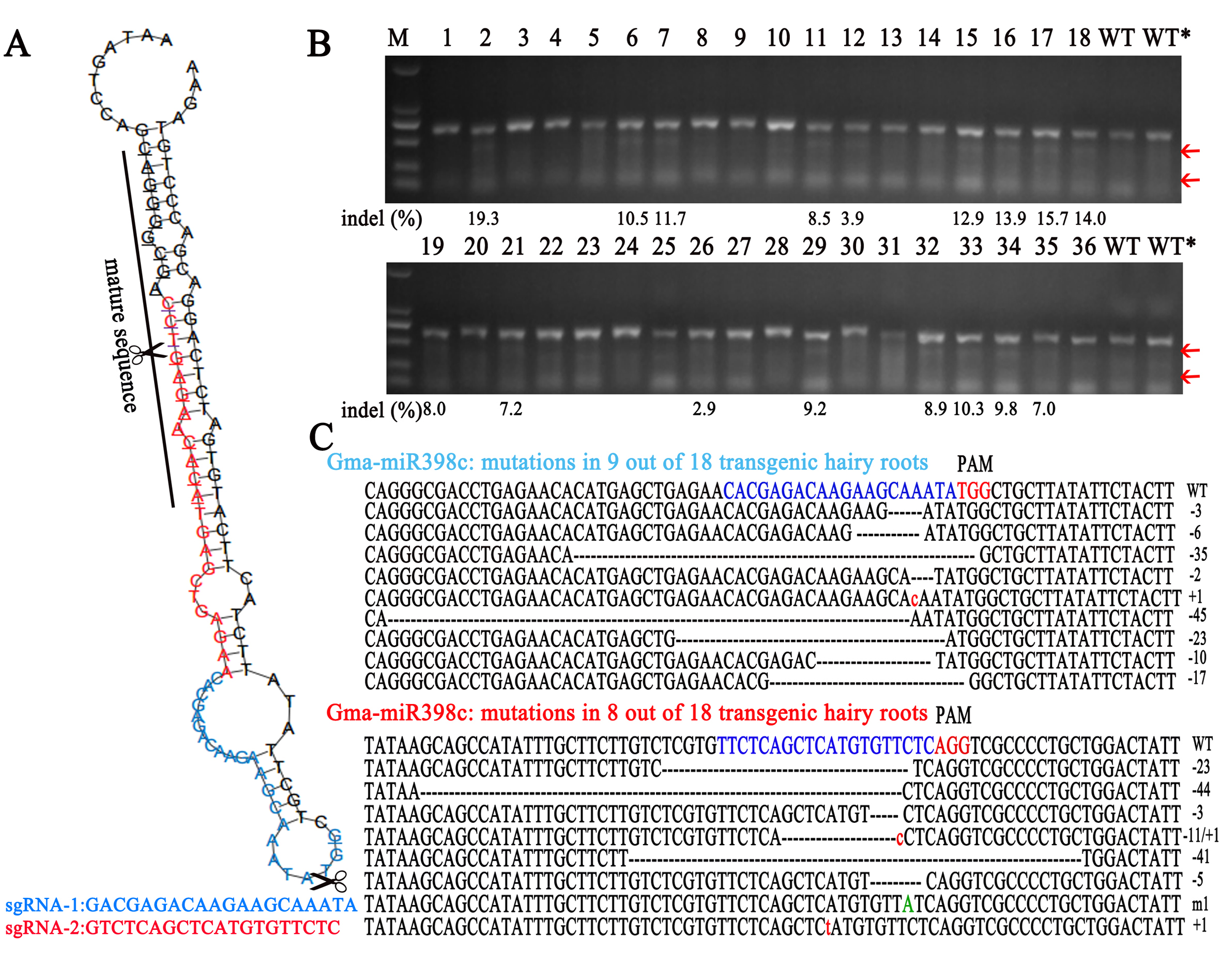
The CRISPR/Cas9 vector was obtained from Biogle (Cat#BGK041, Hangzhou, China). Using the web-based tool CRISPR-P (http://crispr.hzau.edu.cn/), sgRNA-1 and sgRNA-2 sequences were designed on the hairpin and mature sequences of gma-MIR398c, respectively. The sequences of sgRNA-1 and sgRNA-2 with their specific primers were cloned into the CRISPR/Cas9 vector including the soybean U6-specific promoter (pCas9-U6-sgRNA) as a knockout vector (KO-1 and KO-2) (Table S7). The pCAMBIA3301-miR398c vector was used as an overexpression vector. The hypocotyls of 5-day-old soybean seedlings were injected with Agrobacterium rhizogenes (K599) harboring the pCas9-U6-sgRNAs or pCAMBIA3301-miR398c plasmid [53]. The seedlings were cultivated in humidity for 6 days, after which the top leaves were removed and the wound sites were covered with vermiculite for about 15 days until hairy roots about 10 cm in length had developed (Figure S8). The seedlings were transferred to 1/2 Hoagland’s nutrient solution for recovery after the original roots were excised. Seedlings of uniform growth with hairy roots were subjected to 6% PEG 8000 for 48 h to induce simulated drought stress. A total of ten independent replicates were performed.
CRISPR/Cas9 activity assessment
To detect the capacity and efficiency of CRISPR/Cas9 for editing the precursor sequence of gma-miR398c, sgRNA-1 and sgRNA-2 of different regions were inserted in the CRISPR/Cas9 vector (Figure S9A). PCR primers for genomic DNA from transgenic roots were designed to amplify about 705 bp containing the target site (Table S7). The PCR products were denatured and renatured to produce heteroduplexes. Given the absence of an appropriate restriction enzyme site in gma-MIR398c, the T7E1 (E001L, ViewSolid, Beijing, China) assay was performed to detect CRISPR/Cas9-induced mutations. The mutant sites were identified by subcloning and sequencing. To calculate insertion/deletion (indel) frequencies in accordance with the formula: indel (%) = 100 × a / (a + b + c), where a is the intensity of the undigested band, and b and c are the intensities of the other bands, we measured the gel band intensities with ImageJ [54]. We detected the capacity and efficiency of CRISPR/Cas9 for editing gma-MIR398c and identified mutants in 9 and 8 out of 18 independent transgenic hairy roots with indel frequencies ranging from 3.9% to 19.3% and from 2.9% to 10.3%, respectively (Figure S9B). To subclone and sequence the positive PCR products, the indels of mutants at the target sites were conformed (Figure S9C).
Relative electrolyte leakage and superoxide free radical (O2.−) assay
After stress treatment for 48 h, the second trifoliate leaf from the shoot apex of control and treated seedlings was used for a relative electrolyte leakage assay. The leaves of 10 seedlings for each treatment were vacuum-infiltrated for 45 min in water, and placed for 1 h at 25°C. The conductivity (R1) was measured. The leaves were then autoclaved for 15 min and shaken until water dropped to 25°C. Relative electrolyte leakage (%) was calculated as the ratio R1/R2. The production of O2.− was analyzed following the method of Elstner and Heupel [55].
Stomatal aperture measurement
The composite plants were treated with 6% PEG 8000 for 3 h, and plants were placed in 1/2 Hoagland’s nutrient solution as a control. The stoma of the second trifoliate leaf from the shoot apex of control and treated plants was observed with a Leica TCS-SPE confocal microscope.
Data analysis
Data were subjected to analysis of variance. Student’s t-test was used to assess the significance of differences between the means of two datasets (***P < 0.001, **P < 0.01 and *P < 0.05). The data are presented herein as the mean ± standard deviation.
Online data deposition
The sRNA-seq, degradome-seq, and mRNA-seq raw data have been deposited in the NCBI database SRA (PRJNA407016) and the SRR accession numbers are SRR6122987 to SRR6123012.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}