A. Biology methods.
Bacterial Strains, Plasmids, and Media. The strains used in this work are summarized in Table S1. All bacteria were routinely grown at 25 °C in Tryptic Soy Agar or Broth (Pronadisa) supplemented with NaCl (Thermo) up to 1% (TSA-1 or TSB-1). For assays under iron-limiting conditions, the strains were cultured in M9 minimal medium32 supplemented with 0.2% with Casamino Acids (Difco) resulting into CM9 medium. Iron restriction was induced in the CM9 medium by the addition of ethylenediamine-di(o-hydroxyphenylacetic acid) (EDDHA)(Sigma-Aldrich) at the suitable concentration. EDDHA stock solution was prepared dissolving 1 g in 15 ml of NaOH 1N and adjusting the pH to 9.0 with HCl 37% in a final volume of 20 ml.
Growth under Iron-Limiting Conditions. The characterization of biological activity of amonabactins, analogues and probes were carried out in CM9 with the addition of EDDHA 5 µM. The EDDHA minimal inhibitory concentration (MIC) was determined by growing the strains in a gradient of EDDHA (0.5 to 10 µM). Strains VT45.1ΔentB, which carries the amonabactin receptor (FstC) active, and VT45.1ΔentBΔfstC, with FstC inactivated, were cultured in TSB-1 at 25 °C with shaking until an OD600 of 0.5 (mid log phase) and inoculated at a final concentration of 1:40 into wells of a 96-well microtiter plate, containing 200 µl medium per well. Siderophores, analogues or probes were included into the pertinent well at the appropriate concentration (19.5, 13, 6.5, 3.25 or 1.62 µM). The compounds stocks were prepared in a methanol:milliQ-water (1:1) solution at 1.3 mM and stored at -20 °C. Microplates were incubated for 18 h at 25 °C into an iMACK Microplate reader (Bio-Rad) taking measurements every 30 min. All the conditions were assayed in triplicate in each experiment and three independent experiments were performed. All assays incorporated the suitable control wells: media without the addition of siderophore/analogue/probe, media supplemented with FeCl3 10 µM and media without bacteria. Student´s test was performed to analyze the statistical differences between the different conditions.
Fluorescence assays. Sample preparation for microscopy was performed in CM9 medium under weak iron restriction (EDDHA 1 µM). The strains tested were the VT45.1ΔentB, VT45.1ΔentBΔfstC and V. anguillarum RV22. The bacteria were cultured with shaking in TSB-1 at 25 °C until an OD600 of 0.5 (mid log phase) and inoculated into 5 ml of CM9 at a final concentration of 1:40. Probes were added at a concentration of 6.5 µM, then the bacteria were incubated for 12 h at 25 °C with shaking at 150 rpm. When the cultures reached an OD600 of approximately 0.85, 1 ml of the cultures was collected, centrifuged for 3 min at 8000 rpm and the cells were washed by resuspending them in cold (4 °C) PBS. This washing procedure was repeated three times to eliminate residual probe from the medium. After centrifugation, the cells were fixed for 15 min at 4 °C in 1 ml of PBS with 2% p-formaldehyde (Sigma-Aldrich). Finally, the cells were washed twice and visualized at the fluorescence microscope. Imaging was performed on a Confocal Microscope A1R (Nikon) using a 100x oil immersion objective lens. The filter set was the G2A (Ex. 560 nm, Em. 575-615 nm).
B. Chemistry methods.
General information and Procedures. Nuclear magnetic resonance spectra (proton and carbon) were recorded on Bruker 300, 400 and 500 Advance spectrometers at the University of A Coruña, using CDCl3 and CD3OD as the solvents and internal standards. Multiplicities of 13C signals were obtained by DEPT. Medium-pressure chromatographic separations were carried out on silica gel 60 (230–400 mesh). LREIMS and HRESIMS were measured on Applied Biosystems QSTAR Elite. HPLC separations were carried out on an Agilent HP1100 liquid chromatography system equipped with a solvent degasser, quaternary pump, and an UV detector (Agilent Technologies, Waldbronn, Germany). In the HPLC separations, a Discovery® column HS F5 (100x4.6 mm, 5 µm) was used.
All moisture-sensitive reactions were carried out under an atmosphere of argon in flame-dried glassware closed by rubber septum, unless otherwise noted. Solvents were distilled prior to use under argon atmosphere and dried according to standard procedures. Solutions and solvents were added via syringe or cannula. Thin layer chromatography (TLC) was performed using silica gel GF-254 Merck, spots were revealed employing UV light (254 nm) and/or by heating the plate pre-treated with an ethanolic solution of phosphomolybdic acid, a solution of cerium sulphate or a solution of ninhydrin in BuOH-AcOH-H2O. CRYOCOOL apparatus was used for low-temperature reactions.
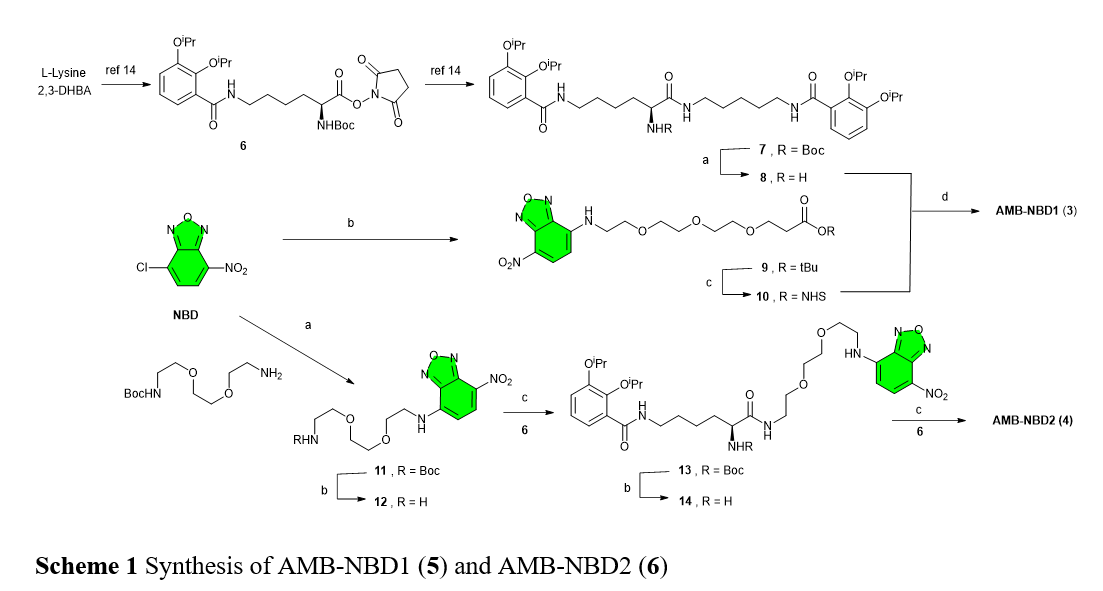
Synthesis of intermediates 8, 9 and 22 are described in previous work.
Synthesis of AMB-NBD1 (5).
Synthesis of 10: A solution of 9 (50 mg, 0.064 mmol) in 5 mL of a mixture of TFA/CH2Cl2 (1:9) was stirred at room temperature for 90 min. Then, the mixture was concentrated under reduced pressure to obtain 10 (42 mg, 0.063, quant.) as a white solid. It was used in next step without characterization.
Synthesis of 11: To a solution of 4-chloro-7-nitrobenzofurazan (200 mg, 1.00 mmol) and cesium carbonate (386 mg, 2.00 mmol) in anhydrous CH3CN (5 mL), was added a solution of tert-butyl 12-amino-4,7,10-trioxadodecanoate (278 mg, 1.00 mmol) in anhydrous CH3CN (5 mL), and the mixture was stirred overnight at room temperature. Then, the reaction was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography eluting with MeOH/CH2Cl2 (4:96) to give 11 (315 mg, 0.72 mmol, 72% yield), as a yellow oil. 1H-NMR (300 MHz, CDCl3): δ 8.46 (d, 1H, H-5); 7,21 (s, 1H, NH); 6.18 (d, 1H, H-6); 3.87 (t, 2H, H-13); 3.67 (m, 12H, H-7-12); 2.48 (t, 2H, H-14); 1.42 (s, 9H, tBu). 13C-NMR (75 MHz, CDCl3): δ 171.1 (C-O, C-15); 144.4 (C, C-3/C-2); 144.1 (C, C-1); 136.6 (CH, C-5); 123.8 (C, C-4); 98.9 (CH, C-6); 80.7 (C, tBu); 70.8-70.5 (CH2, C-9-12); 68.3 (CH2, C-8); 67.0 (CH2, C-13); 44.0 (CH2, C-7); 36.4 (CH2, C-14); 28.2 (CH3, tBu). HRMS (ESI+)m/z: [M+Na]+ calcd. for C19H28N4O8Na: 463.1799; found: 463.1800.
Synthesis of 12: A solution of 11 (100 mg, 0.21 mmol) in TFA/CH2Cl2 (1:9, 5 mL) was stirred for 90 min. After the reaction was complete as monitored by TLC, the mixture was concentrated under reduced pressure to obtain a yellow oil. Then, it was added NHS (47 mg, 0.42 mmol) and EDC·HCl (60 mg, 0.31 mmol), dissolved in anhydrous CH2Cl2 (6 mL), and the mixture was stirred at room temperature overnight. The reaction was washed with water and brine, dried with MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography eluting with MeOH/CH2Cl2 (5:95) to give 12 (80 mg, 0.17, 80%) as a yellow oil. 1H-NMR (400 MHz, CDCl3): δ 8.45 (d, 1H, H-5); 7.43 (s, 1H, NH); 6.19 (d, 1H, H-6); 3.87 (t, 2H, H-13); 3.69 (m, 12H, H-7-12); 2.74 (s, 4H, NHS); 2.60 (t, 2H, H-14). 13C-NMR (100 MHz, CDCl3): δ 175.3 (C-O, C-15); 172.5 (C-O, NHS); 144.5-144.0 (C, C-1-3); 136.7 (CH, C-5); 123.3 (C, C-4); 98.9 (CH, C-6); 70.6-70.2 (CH2, C-9-12); 68.2 (CH2, C-8); 66.3 (CH2, C-13); 43.9 (CH2, C-7); 34.6 (CH2, C-14); 25.4 (CH2, NHS). HRMS (ESI+) m/z: [M+H]+ calcd. for C19H24N5O10: 482.1517; found: 482.1521.
Synthesis of 13: To a solution of 12 (50 mg, 0.074 mmol) in CH2Cl2 (5 mL), was added a solution of 10 (42 mg, 0.082 mmol) in CH2Cl2 (5 mL) and DIPEA (27 µL, 0.150 mmol), and the mixture was stirred at room temperature for 2 h. Then, it was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography eluting with MeOH/CH2Cl2 (5%), to obtain 13 (45 mg, 0.043 mmol, 58%) as a yellow oil. 1H-NMR (400 MHz, CDCl3): δ 8.41 (d, 1H, H-33); 8.09 (2t, 2H, NH-7/19); 7.60 (m, 2H, H-6); 7.14 (d, 1H, NH-20); 7.09-6.90 (m, 5H, H-4/H-5/NH-29); 6.79 (t, 1H, NH-13); 6.17 (d, 1H, H-34); 4.64 (2hept, 2H, iPr); 4.52 (2hept, 2H, iPr); 4.37 (m, 1H, H-12); 3.83 (t, 2H, H-22); 3.65 (m, 12H, H-23-28); 3.40 (m, 4H, H-8/H-18); 3.20 (m, 2H, H-14); 2.48 (m, 2H, H-21); 1.90-1.37 (m, 12H, H-9-11/H-15-17); 1.33 (d, 12H, iPr); 1.26 (d, 12H, iPr). 13C-NMR (100 MHz, CDCl3): δ 171.9 (2C-O, C-13/C-20); 166.2 (2C-O, C-7/C-19); 150.9 (2C, C-2); 146.0 (2C, C-2); 144.4 (C, C-29-31); 136.7 (CH, C-33); 128.4 (2C, C-1); 123.8 (2CH, C-4); 123.7 (C, C-32); 122.7 (2CH, C-5); 118.3 (2CH, C-6); 98.6 (CH, C-34); 76.4 (2CH, iPr); 71.2 (2CH, iPr); 70.7-70.4 (CH2, C-23-26); 68.4 (CH2, C-27); 67.4 (CH2, C-22); 53.4 (CH, C-12); 44.0 (CH2, C-28); 39.5 (CH2, C-8); 39.3 (CH2, C-18); 38.9 (CH2, C-14); 37.0 (CH2, C-21); 31.3 (CH2, C-11); 29.8 (CH2, C-9); 29.5 (2CH2, C-15/C-17); 24.3 (CH2, C-16); 22.9 (CH2, C-10); 22.5 (CH3, iPr); 22.2 (CH3, iPr). HRMS (ESI+) m/z: [M+H]+ calcd. for C52H77N8O14: 1037.5553; found: 1037.5559. [α]D25= +34.5 (c = 0.14, CHCl3).
Synthesis of AMB-NBD1 (5): To a solution of 13 (35 mg, 0.034 mmol) in anhydrous CH2Cl2 (5 mL) at -78°C, was added BCl3 (340 µL, 1M in CH2Cl2), and the mixture was stirred overnight at -40°C. Then, 5 mL of water was added to quench the reaction and concentrated under reduced pressure. The residue was purified by HPLC using a Discovery HS F5 (100 × 4.6 mm, 5 μm) column with a mobile phase consisting on a gradient of 50% CH3CN to 100% in H2O (v/v), each containing 0.1% TFA, for 15 min, at a flow rate of 2 mL/min (injected volume 1 mL; detection 254 nm, retention time 9 min), to give 5 (16 mg, 0.018 mmol, 55%) as an orange oil. 1H-NMR (400 MHz, CD3OD): δ 8.47 (d, 1H, H-33); 7.20 (2dd, 2H, H-6); 6.91 (2dd, 2H, H-4); 6.70 (2t, 2H, H-5); 6.37 (d, 1H, H-34); 4.30 (dd, 1H, H-12); 3.83 (t, 2H, H-22); 3.80-3.50 (m, 14H, H-22-28); 3.38 (m, 4H, H-8/H-18); 3.20 (m, 2H, H-14); 2.47 (t, 2H, H-21); 1.90-1.30 (m, 12H, H-9-11/H-15-17). 13C-NMR (100 MHz, CD3OD): δ 172.8 (C-O, C-20); 172.6 (C-O, C-13); 170.0 (2C-O, C-7/C-19); 148.8 (2C, C-2); 145.9 (2C, C-2); 144.2 (C, C-29-31); 137.0 (CH, C-33); 121.8 (2C, C-1); 118.1 (4CH, C-4/C-5); 117.2 (C, C-32); 115.3 (2CH, C-6); 98.8 (CH, C-34); 70.2-69.9 (CH2, C-23-26); 68.4 (CH2, C-27); 66.8 (CH2, C-22); 53.4 (CH, C-12); 43.4 (CH2, C-28); 38.9-38.7 (3CH2, C-8/C-14/C-18); 36.1 (CH2, C-21); 31.5 (CH2, C-11); 28.6 (3CH2, C-9/C-15/C-17); 23.8 (CH2, C-16); 22.8 (CH2, C-10). HRMS (ESI+) m/z: [M+Na]+ calcd. for C390H53N8O14Na: 891.3495; found: 891.3506. [α]D25= +29.1 (c = 0.11, CH3OH).
Synthesis of AMB-NBD2 (4).
Synthesis of 14: To a solution of 4-chloro-7-nitrobenzofurazan (40 mg, 0.20 mmol) and cesium carbonate (130 mg, 0.40 mmol) in anhydrous CH3CN (5 mL), was added a solution of tert-butyl 12-amino-4,7,10-trioxadodecanoate (50 mg, 0.20 mmol) in anhydrous CH3CN (5 mL), and the mixture was stirred overnight at room temperature. Then, the reaction was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography eluting with EtOAc/Hex (80%) to give 14 (56 mg, 0.14 mmol, 68%), as a yellow oil. 1H-NMR (300 MHz, CDCl3): δ 8.42 (d, 1H, H-5); 7,13 (s, 1H, NH); 5.05 (s, 1H, NH-Boc); 6.18 (d, 1H, H-6); 3.85 (t, 2H, H-11); 3.68 (m, 6H, H-8-10); 3.56 (t, 2H, H-7); 3.32 (c,2H, H-12); 1.39 (s, 9H, Boc). 13C-NMR (75 MHz, CDCl3): δ 156.1 (C-O, Boc); 144.2 (C, C-3/C-2); 143.9 (C, C-1); 136.5 (CH, C-5); 123.6 (C, C-4); 98.9 (CH, C-6); 79.3 (C, Boc); 70.5-70.2 (CH2, C-9-11); 68.2 (CH2, C-8); 43.7 (CH2, C-7); 40.3 (CH2, C-12); 28.4 (CH3, Boc). HRMS (FAB+) m/z: [M+H]+ calcd. for C17H26N5O7: 412.1827; found: 412.1852.
Synthesis of 15: A solution of 14 (50 mg, 0.12 mmol) in TFA: CH2Cl2 (1:9, 5 mL) was stirred for 90 min. After the reaction was complete as monitored by TLC, the mixture was concentrated under reduced pressure to obtain 15 (36 mg, quant.) as a yellow oil. It was used in next step without characterization.
Synthesis of 16: To a solution of 8 (70 mg, 0.12 mmol) in CH2Cl2 (5 mL), was added a solution of 15 (36 mg, 0.11 mmol) in CH2Cl2 (5 mL) and DIPEA (35 µL, 0.200 mmol), and the mixture was stirred at room temperature for 2 h. Then, it was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography eluting with MeOH/CH2Cl2 (5%), to obtain 16 (72 mg, 0.095 mmol, 82%) as a yellow oil. 1H-NMR (300 MHz, CD3Cl): δ 8.41 (d, 1H, H-24); 8.14 (t, 1H, NH-7); 7.62 (dd, 1H, H-6); 6.99 (m, 2H, H-5/H-4); 6.74 (t, 1H, NH-13); 6.18 (d, 1H, H-25); 5.44 (d, 1H, NH-Boc); 4.65 (hept, 1H, iPr); 4.53 (hept, 1H, iPr); 4.04 (c, 1H, H-12); 3.85 (t, 2H, H-15); 3.75-3.30 (m, 12H, H-14/H-16-19); 1.95-1.55 (m, 6H, H-9-11); 1.40 (s, 9H, Boc); 1.34 (d, 6H, iPr); 1.27 (d, 6H, iPr). 13C-NMR (75 MHz, CDCl3): δ 172.4 (C-O, C-13); 166.2 (C-O, C-7); 155.8 (C-O, Boc); 150.7 (C, C-2); 146.0 (C, C-2); 144.4 (C, C-20-22); 136.6 (CH, C-24); 128.1 (C, C-1); 123.6 (CH, C-4); 123.1 (C, C-23); 122.6 (CH, C-5); 118.2 (CH, C-6); 98.7 (CH, C-25); 79.7 (C, Boc); 76.3 (CH, iPr); 71.1 (CH, iPr); 70.5-70.3 (CH2, C-16-17); 69.7 (CH2, C-18); 68.3 (CH2, C-15); 54.6 (CH, C-12); 43.8 (CH2, C-19); 39.1 (CH2, C-8); 38.7 (CH2, C-14); 31.8 (CH2, C-11); 29.4 (CH2, C-9); 28.3 (CH3, Boc); 22.7 (CH2, C-10); 22.3 (CH3, iPr); 22.0 (CH3, iPr). HRMS (FAB+) m/z: [M+H]+ calcd. for C36H54N7O11: 760.3876; found: 760.3860. [α]D25= +35.0 (c = 0.14, CHCl3).
Synthesis of 17: A solution of 16 (70 mg, 0.092 mmol) in TFA: CH2Cl2 (1:9, 5 mL) was stirred for 90 min. After the reaction was complete as monitored by TLC, the mixture was concentrated under reduced pressure to obtain 17 (60 mg, 0.091 mmol, quant.) as a yellow oil. It was used in next step without characterization.
Synthesis of 18: To a solution of 8 (55 mg, 0.097 mmol) in CH2Cl2 (5 mL), was added a solution of 17 (60 mg, 0.091 mmol) in CH2Cl2 (5 mL) and DIPEA (35 µL, 0.200 mmol), and the mixture was stirred at room temperature for 2 h. Then, it was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography eluting with MeOH/CH2Cl2 (4%), to obtain 18 (62 mg, 0.056 mmol, 62%) as a yellow oil. 1H-NMR (300 MHz, CD3Cl):δ 8.45 (d, 1H, H-30); 8.12 (m, 2H, NH-7); 7.65 (m, 2H, H-6); 7.01 (m, 6H, H-5/H-4/NH-13/19); 6.14 (m, 2H, H-31/NH-Boc); 4.65 (2hept, 2H, iPr); 4.54 (2hept, 2H, iPr); 4.35 (m, 1H, H-14); 4.01 (m, 1H, H-12); 3.87 (m, 2H, H-21); 3.75-3.50 (m, 8H, H-22-25); 3.45-3.20 (m, 6H, H-8/H-18/H-20); 2.05-1.50 (m, 12H, H-9-11/H-15-18); 1.44 (s, 9H, Boc); 1.34 (d, 12H, iPr); 1.27 (d, 12H, iPr). 13C-NMR (75 MHz, CDCl3): δ 173.2 (C-O, C-19); 171.7 (C-O, C-13); 166.3 (2C-O, C-7); 156.5 (C-O, Boc); 150.9 (2C, C-2); 146.0 (2C, C-2); 144.3 (C, C-26-28); 136.7 (CH, C-30); 128.2 (2C, C-1); 123.7 (2CH, C-4); 122.6 (2CH, C-5); 122.3 (C, C-29); 118.2 (2CH, C-6); 98.7 (CH, C-31); 80.2 (C, Boc); 76.4 (2CH, iPr); 71.1 (2CH, iPr); 70.6-70.4 (CH2, C-22/C-23); 69.7 (CH2, C-24); 68.3 (CH2, C-21); 55.7 (CH, C-14); 53.4 (CH, C-12); 44.1 (CH2, C-25); 39.4 (CH2, C-20); 38.5 (2CH2, C-8/C-14); 31.3 (CH2, C-11); 30.7 (CH2, C-15); 29.7-29.1 (CH2, C-9/C-17); 28.4 (CH3, Boc); 23.1 (CH2, C-16); 22.7 (CH2, C-10); 22.4 (CH3, iPr); 22.1 (CH3, iPr). HRMS (FAB+)m/z: [M+H]+ calcd. for C55H82N9O15: 1108.5925; found: 1108.5971. [α]D25= +10.7 (c = 0.13, CHCl3).
Synthesis of AMB-NBD2 (6): To a solution of 18 (60 mg, 0.054 mmol) in anhydrous CH2Cl2 (5 mL) at -78°C, was added BCl3 (340 µL, 1M in CH2Cl2), and the mixture was stirred overnight at -40°C. Then, 5 mL of water was added to quench the reaction and concentrated under reduced pressure. The residue was purified by HPLC using a Discovery HS F5 (100 × 4.6 mm, 5 μm) column with a mobile phase consisting of a gradient of 10% CH3CN to 100% in H2O (v/v), each containing 0.1% TFA, for 20 min, at a flow rate of 2 mL/min (injected volume 1 mL; detection 254 nm, retention time 18 min), to give 6 (28 mg, 0.033 mmol, 62%) as an orange oil. 1H-NMR (500 MHz, CD3OD): δ 8.45 (d, 1H, H-30); 7.18 (dd, 2H, H-6); 6.90 (m, 2H, H-4); 6.67 (m, 2H, H-5); 6.35 (d, 1H, H-31); 4.33 (t, 1H, H-14); 3.89 (t, 1H, H-12); 3.79 (t, 2H, H-21); 3.71 (m, 2H, H-24); 3.66-3.57 (m, 4H, H-22/H-23); 3.48 (t, 2H, H-25); 3.42-3.20 (m, 6H, H-8/H-18/H-20); 1.94-1.60 (m, 8H, H-9/H-11/H-15/H-17); 1.46 (m, 4H, H-10/H-16). 13C-NMR (125 MHz, CD3OD): δ 173.6 (C-O, C-13); 171.4 (2C-O, C-7); 170.0 (2C-O, C-19); 150.1 (2C, C-2); 147.3 (2C, C-2); 145.8-145.4 (C, C-27-28); 138.4 (CH, C-30); 123.3 (2C, C-1); 119.6 (2CH, C-4); 118.6 (2C, C-5); 117.2 (C, C-29); 116.7 (2CH, C-6); 114.9 (C, C-26); 100.1 (CH, C-31); 71.6-71.3 (CH2, C-22/C-23); 70.5 (CH2, C-24); 69.8 (CH2, C-21); 54.9 (CH, C-14); 54.2 (CH, C-12); 44.7 (CH2, C-25); 40.3-40.0 (3CH2, C-8/C-18/C-20); 32.9 (CH2, C-11); 32.4 (CH2, C-15); 30.1 (2CH2, C-9/C-17); 24.1 (CH2, C-16); 23.1 (CH2, C-10). HRMS (ESI+) m/z: [M+H]+ calcd. for C38H50N9O13: 840.3523; found: 840.3526. [α]D25= +6.9 (c = 0.10, CH3OH).
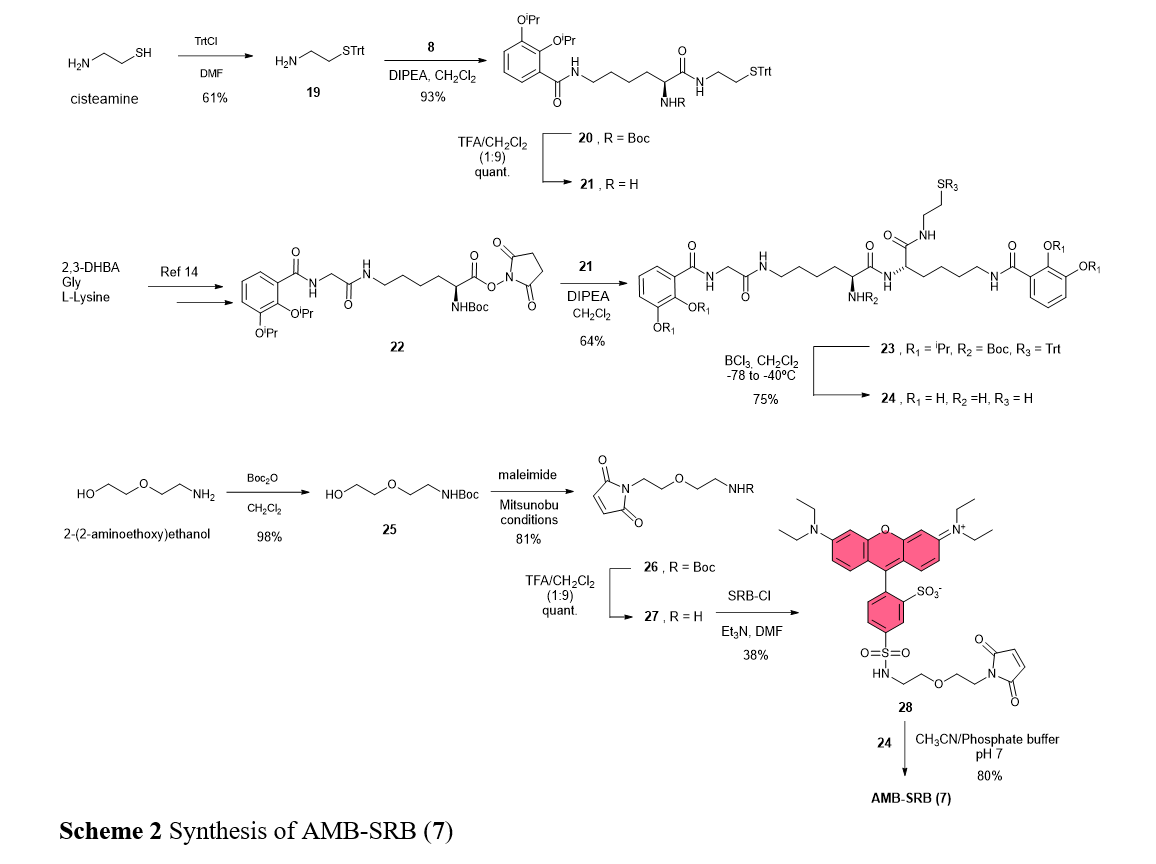
Synthesis of AMB-SRB (7).
Synthesis of 19: A solution of cysteine (200 mg, 2.59 mmol) and trityl chloride (725 mg, 2.60 mmol) in anhydrous DMF (10 mL) was stirred at room temperature overnight. Then, 20 mL of water was added, and the mixture was extracted with EtOAc (2x30mL), washed with water and brine, dried with MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography eluting with MeOH:CH2Cl2 (4:96) to obtain 17 (505 mg, 1.58 mmol, 61%) as a yellow oil. 1H-NMR (300 MHz, CDCl3): δ 7.44 (d, 6H, Trt); 7.26 (m, 9H, Trt); 2.58 (t, 2H, CH2-NH2); 2.36 (t, 2H, CH2-STrt); 2.03 (s, 2H, NH2). 13C-NMR (75 MHz, CDCl3): δ 145.0 (C, Trt); 129.7 (CH, Trt); 128.0 (CH, Trt); 126.8 (CH, Trt); 66.7 (C, Trt); 40.9 (CH2-NH2); 35.8 (CH2-STrt). HRMS (ESI+)m/z: [M+H]+ calcd. for C21H22NS: 320.1467; found: 320.1466.
Syntesis of 20: To a solution of 8 (180 mg, 0.32 mmol) in CH2Cl2 (5 mL) was added a solution of 19 (110 mg, 0.34 mmol) in CH2Cl2 (5 mL) and DIPEA (120 μL, 0.69 mmol), and the mixture was stirred at room temperature for 4 h. Then, it was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography eluting with hexane:EtOAc (3:2) to obtain 20 (228 mg, 0.30 mmol, 93%) as a yellow oil. 1H-NMR (400 MHz, CDCl3): δ 8.07 (t, 1H, NH-7); 7.68 (dd, 1H, H-6); 7.40 (d, 6H, Trt); 7.27 (t, 6H, Trt); 7.20 (t, 3H, Trt); 7.06 (t, 1H, H-5); 7.00 (dd, 1H, H-4); 6.27 (t, 1H, NH-13); 5.21 (d, 1H, NH-Boc); 4.66 (hept, 1H, iPr); 4.55 (hept, 1H, iPr); 3.97 (m, 1H, H12); 3.42 (m, 2H, H-8); 3.05 (c, 2H, H-14); 2.39 (m, 2H, 15); 1.85 (m, 2H, H-11); 1.61 (m, 2H, H-9); 1.39 (s, 9H, Boc); 1.36 (m, 8H, H-9/iPr); 1.28 (d, 6H, iPr). 13C-NMR (100 MHz, CDCl3): δ 171.9 (C-O, C-13); 166.0 (C-O, C-7); 155.7 (C-O, Boc); 150.8 (C, C-2); 146.0 (C, C-3); 144.7 (C, Trt); 129.6 (CH, Trt); 128.4 (C, C-1); 128.0 (CH, Trt); 126.8 (CH, Trt); 123,7 (CH, C-4); 123.0 (CH, C-5); 118.3 (CH, C-6); 79.9 (C, Boc); 76.3 (CH, iPr); 71.1 (CH, iPr); 66.8 (C, Trt); 54.5 (CH, C-12); 38.8 (CH2, C-14); 38.2 (CH2, C-8); 31.8 (CH2, C-15); 29.7 (CH2, C-11); 29.5 (CH2, C-9); 28.3 (CH3, Boc); 22.8 (CH2, C-10); 22.4 (CH3, iPr); 22.1(CH3, iPr). HRMS (ESI+) m/z: [M+H]+ calcd. for C45H58N3O6S: 768.4041; found: 768.4045. [α]D25= +32.3 (c = 0.17, CHCl3).
Synthesis of 21: A solution of 20 (220 mg, 0.29 mmol) in 5 mL of a mixture of TFA/CH2Cl2 (1:9) was stirred at room temperature for 90 min. Then, the mixture was concentrated under reduced pressure to obtain 21 (190 mg, 0.29 mmol, quant.) as a yellow oil. It was used in next step without characterization.
Synthesis of 23: To a solution of 22 (180 mg, 0.29 mmol) in CH2Cl2 (5 mL) was added a solution of 21 (200 mg, 0.30 mmol) in CH2Cl2 (5 mL) and DIPEA (100 μL, 0.58 mmol), and the mixture was stirred at room temperature for 4 h. Then, it was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography eluting with EtOAc to obtain 23 (213 mg, 0.18 mmol, 64%) as a yellow oil. 1H-NMR (400 MHz, CDCl3): δ 8.82 (t, 1H, NH-21); 8.08 (t, 1H, NH-7´); 7.64 (dd, 2H, H-6); 7.38 (d, 6H, Trt); 7.25 (t, 6H, Trt); 7.18 (t, 4H, Trt/NH-15); 7.07 (t, 2H, H-5); 7.00 (m, 3H, H-4/NH-7); 6.84 (t, 1H, NH-9); 5.63 (d, 1H, NH-Boc); 4.69 (2hept, 2H, iPr); 4.54 (2hept, 2H, iPr); 4.32 (m, 1H, 16); 4.09 (m, 3H, H-8/H-14); 3.37 (m, 2H, H-20); 3.20 (c, 2H, H-10); 3.07 (m, 2H, H-22); 2.39 (t, 2H, H-23); 1.80-1.50 (m, 4H, H-13/H-15/H-17/H-19); 1.39 (s, 9H, Boc); 1.34 (m, 22H, H-11/H-18/iPr); 1.28 (d, 6H, iPr). 13C-NMR (100 MHz, CDCl3): δ 172.6 (C-O, C-21); 171.5 (C-O, C-15); 169.0 (C-O, C-9); 166.2 (C-O, C-7); 156.0 (C-O, Boc); 150.9 (C, C-2); 146.9/146.0 (C, C-3); 144.7 (C, Trt); 129.6 (CH, Trt); 128.4/127.5 (C, C-1); 127.9 (CH, Trt); 126.7 (CH, Trt); 123,8/123.6 (CH, C-4); 122.9/122.7 (CH, C-5); 119.4/118.3 (CH, C-6); 79.9 (C, Boc); 76.6/76.3 (CH, iPr); 71.5/71.1 (CH, iPr); 66.7 (C, Trt); 54.8 (CH, C-16); 53.2 (CH, C-14); 43.9 (CH2, C-8); 39.0 (CH2, C-22);38.7 (CH2, C-20); 38.6 (CH2, C-10); 31.9 (CH2, C-17); 31.7 (CH2, C-23); 31.2 (CH2, C-13); 29.4 (CH2, C-19); 28.7 (CH2, C-11); 28.4 (CH3, Boc); 22.7 (CH2, C-18/C-12); 22.4 (CH3, iPr); 22.1(CH3, iPr). HRMS (ESI+) m/z: [M+Na]+ calcd. for C66H88N6O11SNa: 1195.6124; found: 1195.6124. [α]D25= +19.8 (c = 0.12, CHCl3).
Synthesis of 24: To a solution of 23 (180 mg, 0.15 mmol) in anhydrous CH2Cl2 (5 mL) at −78 °C was added BCl3 (1.5 mL, 1 M in CH2Cl2), and the mixture was stirred overnight at −40 °C. Then, 5 mL of water was added to quench the reaction and concentrated under reduced pressure. The residue was purified by HPLC using a Discovery HS F5 (100 × 4.6 mm, 5 μm) column with a mobile phase consisting of a gradient of 10% CH3CN to 100% in H2O (v/v), each containing 0.1% TFA, for 15 min, at a flow rate of 2 mL/min (injected volume 1 mL; detection 254 nm, retention time 12 min), to give 24 (76 mg, 0.11 mmol, 75%) as a white solid. 1H-NMR (500 MHz, CD3OD): δ 7.23 (dd, 2H, H-6); 6.93 (dd, 2H, H-4); 6.73 (t, 2H, H-5); 4.31 (t, 1H, 16); 4.04 (d, 2H, H-8); 3.86 (t, 1H, 14); 3.39 (m, 2H, H-20); 3.34 (m, 2H, H-10); 3.26 (m, 2H, H-22); 2.58 (t, 2H, 23); 1.81 (m, 4H, H-13/H-17); 1.61 (m, 4H, H-11/H-19); 1.45 (m, 4H, H-12/H-18). 13C-NMR (125 MHz, CD3OD): δ 173.9 (C-O, C-21); 171.8 (C-O, C-7); 171.4 (C-O, C-9); 170.0 (C-O, C-15); 150.0 (C, C-2); 147.3 (C, C-3); 119.8 (CH, C-4); 119.6 (CH, C-5); 118.7/ 119.2 (CH, C-6); 116.,9 (C, C-1); 55.1 (CH, C-16); 54.2 (CH, C-14); 43.8 (CH2, C-8/C-22); 40.0 (CH2, C-20); 39.6 (CH2, C-10); 32.8 (CH2, C-17); 32.2 (CH2, C-13); 30.1 (CH2, C-19); 29.9 (CH2, C-11); 24.5 (CH2, C-23); 24.2 (CH2, C-18); 22.8 (CH2, C-12). HRMS (ESI+) m/z: [M+H]+ calcd. for C30H43N6O9S: 663.2807; found: 663.2796. [α]D25= +11.0 (c = 0.13, CH3OH).
Synthesis of 25: To a solution of 2-(2-aminoethoxy)ethanol (450 µL, 4.49 mmol), and Boc2O (1g, 4.57 mmol) in anhydrous CH2Cl2, was added DIPEA (860 µL, 4.94 mmol), and the mixture was stirred overnight at room temperature. Then, it was concentrated under reduced pressure, and the residue was redisolved in EtOAc (15 mL), washed with water and brine, dried with MgSO4, filtered and concentrated under reduced pressure, to obtain 25 (906 mg, 4.41 mmol, 98%) as a colourless oil. 1H-NMR (300 MHz, CDCl3): δ 5.15 (s, 1H, NH-Boc); 3.70 (t, 2H, H-1); 3.52 (m, 4H, H-2/H-3); 3.29 (c, 2H, H-4); 2.76 (s, 1H, OH); 1.41 (s, 9H, Boc). 13C-NMR (75 MHz, CDCl3): δ 156.2 (C-O, Boc); 79.4 (C, Boc); 72.3 (CH2, C-3); 70.4 (CH2, C-2); 61.7 (CH2, C-1); 40.5 (CH2, C-4); 28.5 (CH3, Boc). HRMS (ESI+) m/z: [M+Na]+ calcd. for C9H19NO4Na: 228.1206; found: 228.1207.
Synthesis of 26: To a solution of PPh3 (716 mg, 2.73 mmol) in anhydrous THF (8 mL) at -78 °C, was added sequentially DEAD (435 µL, 2.73 mmol), a solution of 25 (616 mg, 3.00 mmol) in 4 mL of anhydrous THF, neopentyl alcohol (120 mg, 1.36 mmol), and maleimide (265 mg, 2.73 mmol). After 5 min, the reaction was allowed to reach room temperature, and was stirred overnight. Then, it was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography eluting with hexane:EtOAc (2:1) to obtain 26 (630 mg, 2.21 mmol, 81%) as a white solid.1H-NMR (300 MHz, CDCl3): δ 6.66 (s, 2H, H-1); 4.92 (s, 1H, NH-Boc); 3.70 (t, 2H, H-3); 3.53 (t, 2H, H-4); 3.42 (t, 2H, H-5); 3.18 (c, 2H, H-6); 1.41 (s, 9H, Boc). 13C-NMR (75 MHz, CDCl3): δ 170.7 (C-O, C-2); 155.9 (C-O, Boc); 134.2 (CH, C-1); 79.2 (C, Boc); 69.8 (CH2, C-5); 67.7 (CH2, C-4); 40.3 (CH2, C-6); 37.2 (CH2, C-3); 28.4 (CH3, Boc). HRMS (ESI+) m/z: [M+Na]+ calcd. for C13H20N2O5Na: 307.1264; found: 307.1257.
Synthesis of 27: A solution of 26 (220 mg, 0.29 mmol) in 5 mL of a mixture of TFA/CH2Cl2 (1:9) was stirred at room temperature for 90 min. Then, the mixture was concentrated under reduced pressure to obtain 27 (205 mg, 0.29 mmol, quant.) as a white solid. It was used in next step without characterization.
Synthesis of 28: To a solution of sulforhodamine B acid chloride (30 mg, 0.05 mmol) and 27 (31 mg, 0.01 mmol) in anhydrous DMF (4 mL), was added Et3N (30 µL, 0.02 mmol), and the mixture was stirred overnight at room temperature. Then, it was concentrated under reduced pressure, and the residue was purified by HPLC using a Atlantis dC18 (100 × 10 mm, 5 μm) column with a mobile phase consisting on a gradient of 10% CH3CN to 100% in H2O (v/v), each containing 0.1% TFA, for 15 min, at a flow rate of 2 mL/min (injected volume 1 mL; detection 254 nm, retention time 10 min), to give 28 (28 mg, 0.04 mmol, 38%) as a purple solid. 1H-NMR (500 MHz, CD3OD): δ 8.67 (d, 1H, H-8); 8.13 (dd, 1H, H-12); 7.54 (d, 1H, H-11); 7.14 (d, 2H, H-19); 7.04 (dd, 2H, H-18); 6.96 (d, 2H, H-16); 6.84 (s, 2H, H-1); 3.69 (m, 10H, H-20/H-3); 3.57 (t, 2H, H-4); 3.53 (t, 2H, H-5); 3.19 (t, 2H, H-6); 1.32 (t, 12H, H-21). 13C-NMR (125 MHz, CD3OD): δ 172.5 (C-O, C-2); 159.4 (C, C-14); 157.8 (C, C-13); 157.1 (C, C-17); 147.1 (C, C-7); 144.2 (C, C-9); 135.5 (CH, C-1); 135.4 (C, C-10); 133.7 (CH, C-19); 132.4 (CH, C-11); 129.3 (CH, C-12); 127.6 (CH, C-8); 115.3 (C, C-14); 115.1 (CH, C-18); 96.9 (CH, C-16); 70.2 (CH2, C-5); 69.9 (CH2, C-4); 46.8 (CH2, C-20); 44.0 (CH2, C-6); 38.1 (CH2, C-3); 12.8 (CH3, C-21). HRMS (ESI+) m/z: [M+H]+ calcd. for C₃₅H₄₁N₄O₉S₂: 725.2309; found: 725.2305.
Synthesis of AMB-SRB (7): To asolution of 24 (4.5 mg, 0.007 mmol) in 2 mL of CH3CN:Phosphate buffer (0,1 M, pH=7) (2:1) mixture, was added a solution of 28 (5 mg, 0.007 mmol) in 2 mL of the same mixture, and was stirred at room temperature for 2h. Then, it was concentrated under reduced pressure, and the residue was purified by HPLC using a Discovery HS F5 (100 × 4.6 mm, 5 μm) column with a mobile phase consisting on a gradient of 10% CH3CN to 100% in H2O (v/v), each containing 0.1% TFA, for 15 min, at a flow rate of 2 mL/min (injected volume 1 mL; detection 254 nm, retention time 14 min), to give 7 (7.5 mg, 0.005 mmol, 80%) as a purple solid. 1H-NMR (500 MHz, CD3OD):δ 8.72 (d, 1H, H-33); 8.11 (dd, 1H, H-37); 7.51 (d, 1H, H-36); 7.20 (d, 1H, H-6); 7.09 (m, 3H, H-6/H-44); 6.93 (m, 3H, H-4/H-43); 6.87 (d, 1H, H-4); 6.82 (m, 2H, H-41); 6.67 (t, 1H, H-5); 6.58 (t, 1H, H-5); 4.34 (dd, 1H, H-16); 3.97 (m, 3H, H-8/H-24); 3.90 (dd, 1H, H-14); 3.66 (m, 2H, H-28); 3.60 (m, 10H, H-45/H-29); 3.52 (t, 2H, H-30); 3.44 (t, 2H, H-22); 3.24 (m, 5H, H-10/H-20/H-25); 3.15 (m, 2H, H-31); 3.00 (m, 1H, H-23); 2.79 (m, 1H, H-23); 2.53 (m, 1H, H-25); 1.80 (m, 4H, H-17/H-13); 1.56 (m, 4H, H-19/H-11); 1.45 (m, 4H, H-18/H-12); 1.26 (t, 12H, H-46). 13C-NMR (125 MHz, CD3OD):δ 179.0 (C-O, C-26); 177.1 (C-O, C-27); 173.9 (C-O, C-21); 171.7 (C-O, C-7); 171.5 (C-O, C-9); 170.1 (C-O, C-15); 159.2 (C, C-39); 157.6 (C, C-38); 157.0 (C, C-42); 150.3/150.1 (C, C-2); 147.3 (C, C-3); 147.1 (C, C-32); 143.9 (C, C-34); 135.5 (C, C-35); 133.6 (CH, C-44); 132.6 (CH, C-36); 129.5 (CH, C-37); 127.7 (CH, C-33); 119.8 (CH, C-4); 119.5 (CH, C-5); 119.2/118.7 (CH, C-6); 116.6 (C, C-1º); 115.2 (C, C-39); 115.0 (CH, C-43); 96.9 (CH, C-41); 70.1 (CH2, C-30); 68.1 (CH2, C-29); 55.1 (CH, C-16); 54.3 (CH, C-14); 46.8 (CH2, C-45); 44.2 (CH2, C-31); 43.7 (CH2, C-8); 40.6 (CH, C-24); 40.0 (CH2, C-22); 39.9 (CH2, C-20); 39.6 (CH2, C-10); 39.4 (CH2, C-28); 37.1 (CH2, C-25); 32.8 (CH2, C-17); 32.2 (CH2, C-13); 31.7 (CH2, C-23); 29.9 (CH2, C-19); 29.8 (CH2, C-11); 24.2 (CH2, C-18); 22.9 (CH2, C-12); 12.9 (CH3, C-46). HRMS (ESI+)m/z: [M+H]+ calcd. for C65H83N10O18S3: 1387.5043; found: 1387.5026. [α]D25= +6.7 (c = 0.10, CH3OH).
{kind=link}
{kind=link}