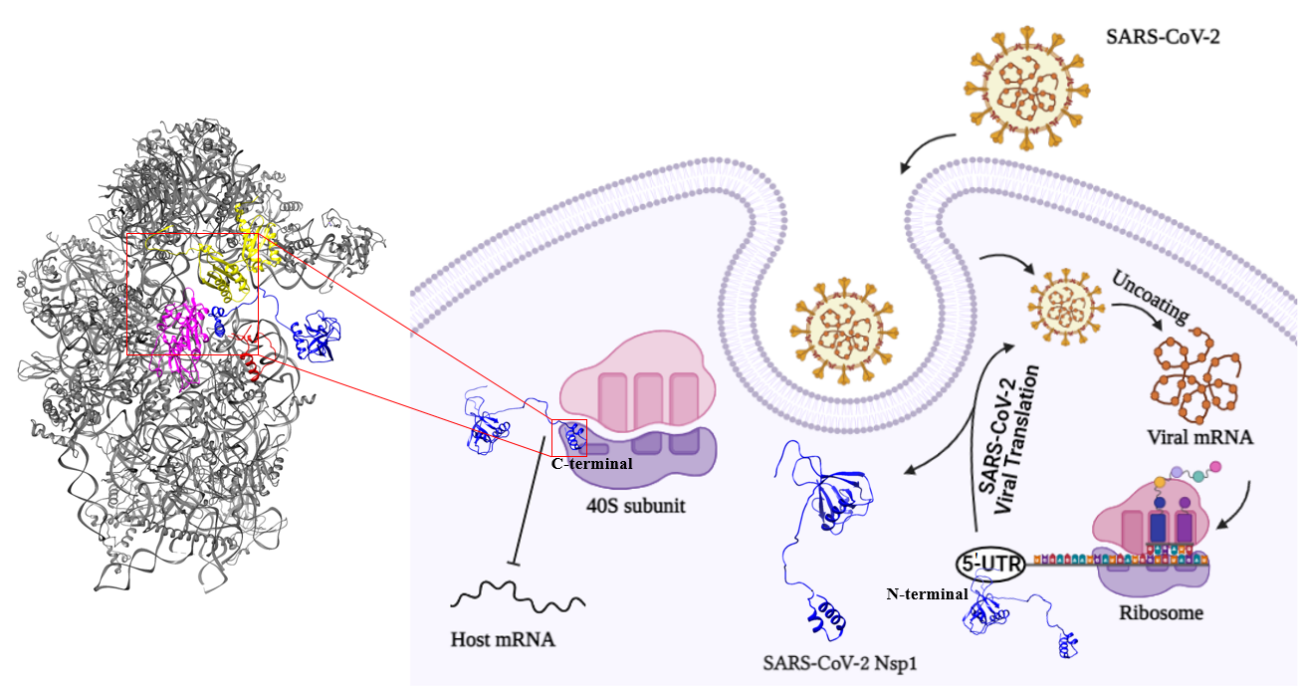
3.1. Sequence and structure alignment
The results of the amino acid sequence alignment revealed that the Nsp1 of SARS-CoV-2 shares 84.4% identity and 91.1% similarity with the Nsp1 of SARS-CoV-1. The tertiary structure of SARS-CoV-2 NSP1 has consisted of regular secondary structural elements arranged sequentially as β1, α1, β2, α2, β3, β4, β5, β6, α3, and α4, which secondary structure is composed of a mixture of parallel and antiparallel β-strands (Fig. 1). The β-strands consist of residues 14 to 20 (β1), 51 to 55(β2), 68 to 73 (β3), 87 to 91 (β4), 104 to 109 (β6), and 117 to 123 (β6), while β5, β1 and β6 are shorter and β3 and β4 are longer than identical β-strands of SARS-CoV-1 Nsp1. Also, NSP1 contains four α-helixes namely α1 (35 to 49), α2 (60 to 63), α3 (153 to 160), and α4 (164–178) that α3 and α4 locate at the C-terminal of protein. Investigation of the Nsp1-ribosomecomplex revealed that the α3 and α4 play a key role in forming this complex. In particular, several specific residues (Y154, E155, D156, F157, Q158, E159, N160, W161, T170, L173, M174, L177, N178) in this region were identified as essential residues involved in the interaction with components of the 40S ribosome subunit (U3 and S2). Most of these residues are found in the list of SARS-CoV-2 Nsp1 mutations. Therefore, examining important residues and understanding their position in protein sequence and structure can provide a novel insight into the effect of mutations on protein function. Furthermore, the Nsp1 has only one cysteine at position 51, which is the first amino acid of β2. This residue is significant because its neighboring residues may mutate and create an extra cysteine. It is suggested that it may lead to form disulfide bond and increase the stability of the protein structure.
3.2. Analysis of Nsp1 complexes and effect of mutations
The complex structure of the SARS-CoV-2 Nsp1 protein and human ribosomal 40S subunit were analyzed. The binding site of Nsp1 to the ribosome contains 26 residues at Nsp1 C-terminal. Among these residues, 17 residues form a helix-turn-helix (HTH) motif and lead to form Nsp1-ribosome complex. HTH interacts simultaneously with different regions of the ribosome called “head region” including uS3 and “body region” including eS30 and uS5. HTH interacts with uS3 by D152, E155, D156, E159, Q158, and N160 residues, as well as this motif involved in interaction with uS5 by Y154, F157, Q158, W161, T170, L173, L177, N178 residues. R175, E176, and G179 residues of HTH motif play a key role in forming Nsp1-ribosome (eS30) complex. We calculated the binding energy (ΔGBinding) for these interactions, which showed that the interaction between the Nsp1 and uS3 is stronger than the other interactions (Table 1).
Table 1
Binding affinity (ΔG) and dissociation constant (Kd) predicted values for the interaction between SARS-CoV-2 Nsp1 and human ribosome.
|
Protein-protein complex
(Viral Nsp1/ribosome)
|
ΔG (kcal/mol)
|
Kd (M) at 37.0 ֯C
|
|
Nsp1-uS3
|
-6.9
|
1.3E-05
|
|
Nsp1-uS5
|
-5.6
|
1.1E-04
|
|
Nsp1-eS30
|
-4.1
|
1.2E-03
|
Substitutions F157S, E159D, L173P and R175H had ΔΔGDestablizing <0 and ΔΔSVib > 0 (Fig. 2), suggesting that they may increase the infectivity of host cells by destabilizing the Nsp1 and increasing its entropy and flexibility. Other mutations may have the opposite effect on Nsp1, such as E159K, T170I, M174K and N178K, which have ΔΔGDestablizing >0 and ΔΔSVib < 0, which stabilize the protein and reduce its entropy (Fig. 2). These substitutions may reduce the rate at which the Nsp1 binds to the host ribosomal subunits and thus reduces the rate of infection.
We identified W161 in the HTH motif as causing a turn in that motif, leading to the helix disruption. This residue can also cause hydrophobic interactions (with F157) and stabilize the protein structure. Several mutations are known for this residue, including W161C, W161S, W161L, and W161R. We found that W161C has ΔΔGDestablizing < -1, which may leads to instability of the protein structure. Mutation F157S can also affect the formation or loss of this hydrophobic interaction, which in turn probably impacts the stability of the Nsp1 in that region.
Investigation of Nsp1-RNA complex have shown that protein can interact with viral RNA through their residues, which include 41E, 47K, 48D, 49G, 50T, 51C, 72K, 75D, 77R, 82G, 96Q, 97T, 99R, 102E, 112G, 113E and 114I (with Hydrogen bond)/ 122L (Hydrophobic interaction)/ 31F (π-stacking) (Fig. 3a). Several residues at the surface interaction of Nsp1-RNA are known to have mutated. Among the most frequent mutations (Table 2), G112 change to C/S and G49 to C result in ΔΔGDestabilizing below zero. These results suggested that the mutation of glycine to cysteine and serine could lead to protein instability, increased flexibility of Nsp1 and increase entropy of this protein. These factors can increase the rate at which Nsp1 binds to viral RNA. D75G and L122F mutations also showed negative values for ΔΔGDestabilizing and ΔΔSVis, indicating that they affect structure stability and thermodynamics parameters (i.e. decrease entropy and protein flexibility). A third group of mutations (D75E and E102K) leads to increase stability, decrease entropy and flexibility of protein, which perhaps reduce protein binding to the viral RNA, thereby reducing infectivity. The last group of mutations includes D48G, which showed positive values for ΔΔGDestabilizing and ΔΔSVis. Based on results of calculations by computational tools, this mutation appears to increase stability and entropy of Nsp1.
Figure 3b shows the surface potential of the Nsp1-RNA complex that indicating the Nsp1 binds to viral RNA through its positive residues in that region.
Some mutations also have effects on intra-atomic interactions of Nsp1 that lead to the elimination and formation of interactions such as van der Waals (yellow), weak Hydrogen bond (orange), and hydrophobic interactions (green), polar (light pink), as shown in Fig. 4.
Table 2
Frequency, ΔΔGDestabilizing and ΔΔSVib are achieved for the mutations of Nsp1 N-ter.
|
Mutation
|
Frequency
|
ΔΔGDestabilizing
(kcal/mol)
|
ΔΔSVib
(kcal.mol− 1.K− 1)
|
|
G112C
|
102
|
-0.800
|
0.032
|
|
G49C
|
118
|
-1.765
|
0.084
|
|
G112S
|
145
|
-1.060
|
0.039
|
|
D75G
|
161
|
-0.244
|
-0.005
|
|
L122F
|
224
|
-0.077
|
-0.811
|
|
I114V
|
232
|
-0.097
|
0.058
|
|
E102K
|
321
|
0.534
|
-0.185
|
|
I114L
|
403
|
-0.164
|
0.072
|
|
D75E
|
990
|
0.848
|
-0.265
|
|
D48G
|
1185
|
0.001
|
0.089
|
3.3. MD simulation
Molecular dynamics (MD) simulations were performed for the wild type and mutant types of SARS-CoV-2 Nsp1 and SARS-CoV-1 Nsp1. The RMSD plot indicates that all proteins reached equilibrium after 100 ns and that the SARS-CoV-1 Nsp1 deviation is less than the SARS-CoV-2 Nsp1 (Fig. 5a). We identified four main deletions in Nsp1 at sites 80–90, 156–158, 158–161 and 175–177. The deletions result in different conformations in Nsp1 C-terminal region (Fig. 6). SARS-CoV-2 Nsp1 (Δ175–177) and Nsp1 (Δ1158−161) follow a pattern similar to SARS-CoV-1 Nsp1 and have less fluctuations than other proteins. This result indicates that the Nsp1 has a lower value of RMSD with the 175–177 and 158–161 deletions, so the mutated proteins are more stable than the wild protein and other mutant protein (Fig. 5a). In contrast, Δ156–158 and Δ80–90 have higher RMSD values than SARS-CoV-2 Nsp1 wild type, which may indicate increasing protein fluctuations and instability.
The SARS-CoV-1 Nsp1 exhibited a lower Rg values compared to the SARS-CoV-2 Nsp1 (Fig. 5b). The difference between Rg of these two proteins is ~ 0.12 nm. Overall, all models of SARS-CoV-2 Nsp1 had various values of Rg that including Nsp1 (Δ156–158) > Nsp1 (wild) > Nsp1 (Δ80−90) > Nsp1 (Δ175−177) > Nsp1 (Δ158−161). Nsp1 (Δ156−158) has more functional movements due to its higher value of Rg and lesser compactness.
The RMS plot was constructed from the 100 ns data in order to understand the deviation of each Nsp1 (Fig. 5c). The RMSF of the backbone for the SARS-CoV-2 Nsp1 displayed more flexible 164 to 170 residues (Lys, His, Ser, Ser, Gly, Val, and Ter), as compared to the SARS-CoV-1 Nsp1. This region corresponds to the C-terminal of the Nsp1 related to the protein interaction with the ribosome through CT, meaning that its flexibility could be related to its function. As shown in Fig. 5c, the RMSF plot of mutant proteins had more fluctuations compared to wild Nsp1 due to their deleted residues. In order to understand the total motion of Nsp1 structures in the phase space, the first two eigenvectors were projected onto it. The results indicate that the motion features characterized by the two eigenvectors are different in the SARS-CoV-2 and SARS-CoV-1 Nsp1 proteins. SARS-CoV-2 Nsp1 show wider motions than SARS-CoV-1 Nsp1, indicating more conformational changes.
As shown in Fig. 5(d-e), the values of RMSD and Rg resulting from the analysis of Nsp1-RNA trajectories after ~ 73 ns MD simulation are 0.6 nm and 1.5 nm, respectively. The RMSF plot of this complex also showed fluctuations in the site of RNA binding to the protein (Fig. 5f).
3.4. Analysis of Network and IDRs of Nsp1
Critical residues at the interface between the SARS-CoV-2 Nsp1 and ribosome subunits in their complex are indicated in the RIN presented (Fig. 7). Key residues were determined by using stress and betweenness as the two main parameters of the local metrics. High betweenness values represent that the node is a mediator of interactions with other nodes and that is probably a key structural residue, while stress is defined as the number of shortest paths passing through a node. The analysis of NSP1 (C-terminal) network revealed crucial residues including F157, V169, T170, and L173. These amino acids were identified as critical residues at the interface between the SARS-CoV-2 NSP1 and ribosome. Furthermore, these residues were known as amino acids that have mutated.
In addition, Table 3 describes the most important residues of the SARS-CoV-2 involved in the interaction between its Nsp1 and ribosome via H-bonds, van der Waals bonds, and π-π stacking interactions. Among these residues, Δ158-161, Δ156-158, Δ175-177, F157S, E159D, L173P and R175H are critical because their mutations could alter the ability of the Nsp1 to bind to ribosome, which could lead to the loss of efficacy of drugs targeting the Nsp1 or competing with it for ribosome.
Table 3
Representation of interaction between different residues of the SARS-CoV-2 Nsp1 and ribosome subunits.
|
Residues of Ribosome
|
Residues of Nsp1
|
Interaction
|
|
(uS5)
105GLU
106VAL
109ILE
109ILE
122THR
124PHE
124PHE
147VAL
147VAL
147VAL
147VAL
151ILE
|
178ASN
177LEU
157PHE
173LEU
161TRP
161TRP
161TRP
154TYR
158GLN
158GLN
161TRP
177LEU
|
VDW
VDW
VDW
VDW
VDW
PIPISTACK
VDW
VDW
HBOND
VDW
VDW
VDW
|
|
(uS3)
115VAL
116ARG
116ARG
116ARG
143ARG
143ARG
143ARG
|
155GLU
152ASP
155GLU
155GLU
155GLU
156ASP
159GLU
|
HBOND
HBOND
HBOND
VDW
HBOND
VDW
VDW
|
|
(eS30)
52LYS
|
175ARG
|
VDW
|
The VSL2 predictor was used to characterize disordered regions of any length and identified accurately the short disordered regions of SARS-CoV-2 NSP1 at 120–180 positions (Fig. 8). PONDR VL3 predictor was applied to determine SARS-CoV-2 NSP1 tendency for disorder. This predictor identified regions of interest at positions 150–180 (Fig. 8). The regions identified as disorder regions are approximately related to the C-terminal protein. It is also suggested that the occurrence of mutations in this region may increase these disorders in the protein. Structural studies revealed that IDRs of virus structure influence viral genome (RNA) adaptive capacity, host-virus interactions, virus-host range, cross-species transmission, and host tropism [37–41]. In antigen selection, IDRs are significantly considered because these disordered regions may induce an undesirable immune response (weak or even completely non-immune responses)[42].
{kind=link}