Genome-wide CRISPR knockout screen identifies genes protective against αS seeding.
To identify genes that regulate αS seeding, we performed a genome-wide screen using a HEK293 αS CFP/YFP biosensor cell line (αS biosensor) (Fig. 1A) [4]. Exogenously applied pre-formed fibrillar αS (αS PFF), but not monomeric αS, seeds the aggregation of soluble intracellular αS resulting in a quantifiable change in FRET efficiency. We clonally expressed spCAS9 in αS biosensors and then infected with a pooled Brunello gRNA library covering 19114 different genes/ 4 gRNA each and 1000 non-targeting controls at a low MOI (< 0.3) for seven days with puromycin selection. The pooled knockdown αS biosensor maintained its normal αS seeding capacity (Figure S1). These biosensors were treated with αS PFF as previously described and flow-sorted into FRET positive and FRET negative groups. DNA was isolated from FRET positive, FRET negative, and the unseeded total cell population. Then next-generation sequencing was performed to identify the guide RNAs (gene knockdowns) represented in each group. This sequence data were analyzed via the MegaCK algorithm in both the FRET positive and negative populations compared with untreated control. 110 genes were enriched in the FRET positive population as compared to the total population, and 43 genes were underrepresented in the FRET negative group versus the total population (FDR < 0.05 and fold change > 2 or < 0.5) (Fig. 1B). These 153 genes were considered “protective” or suppressors of αS seeding in the biosensor line (Fig. 1B-C; Table S1).
Our screen identified genes and pathways previously identified in other screens for αS toxicity. Notably, we identified 15 genes associated with ER-Golgi-endosome trafficking. These included VPS51 and VPS52, which are components of the Golgi-associated retrograde protein (GARP) complex. The GARP complex interacts with Parkinson-associated protein, LRRK2, and deletion of either VPS51 or VPS52 homologs in yeast increases αS accumulation and toxicity [29]. Other modifiers not previously identified in screens include ATP6V0B, ATP6V0C, and ATP6V1A that encode subunits of Vacuolar-type ATPase (V-ATPase). ATP6V0B KD inhibits autophagic degradation and increases αS aggregation [30], and is downregulated in patients with αS inclusions [31]. Pathway analysis identified the enrichment in genes associated with the cellular stress response, such as VCP, SEC61B, and KDELR1. Notably, the ER stress response is upregulated in PD brains and correlates with αS toxicity in multiple model systems [32].
We validated nine candidate suppressors (ATP6V0C, KDELR1, LAMTOR5, RAB35, RABAC1, SEC61B, TMEM147, VCP, and VPS51) using siRNA knockdown in αS biosensors. Following 48 hours of siRNA treatment, αS PFF was added with Lipofectamine, and FRET efficiency was measured 24 hours later. Six candidate suppressors, when knocked down, increased FRET efficiency and included ATP6V0C, VPS51, KDELR1, SEC61B, LAMTOR5, and VCP (Fig. 1D). To further confirm our findings with VCP, we generated a lentiviral vector expressing a VCP specific or control gRNA, infected spCAS9 αS biosensors for 7 days and treated with αS PFF. Similar to that seen with VCP siRNA, FRET efficiency was increased in VCP CRISPR KO αS biosensors (Fig. 1E-F).
VCP inhibition increases α-synuclein seeding efficiency.
As previously reported, αS seeding as measured by FRET is not seen with the application of monomeric αS [4]. Moreover, Lipofectamine is necessary for efficient FRET. The application of “naked” αS PFF to αS biosensors fails to increase FRET after 24 and 48 hours, suggesting that Lipofectamine facilitates entry into the endolysosomal pathway [4]. Consistent with this, the enhanced seeding efficiency and FRET signal following VCP siRNA treatment required the application of αS PFF with Lipofectamine and did not occur with monomeric αS or when αS PFF were added without Lipofectamine (Fig. 2A).
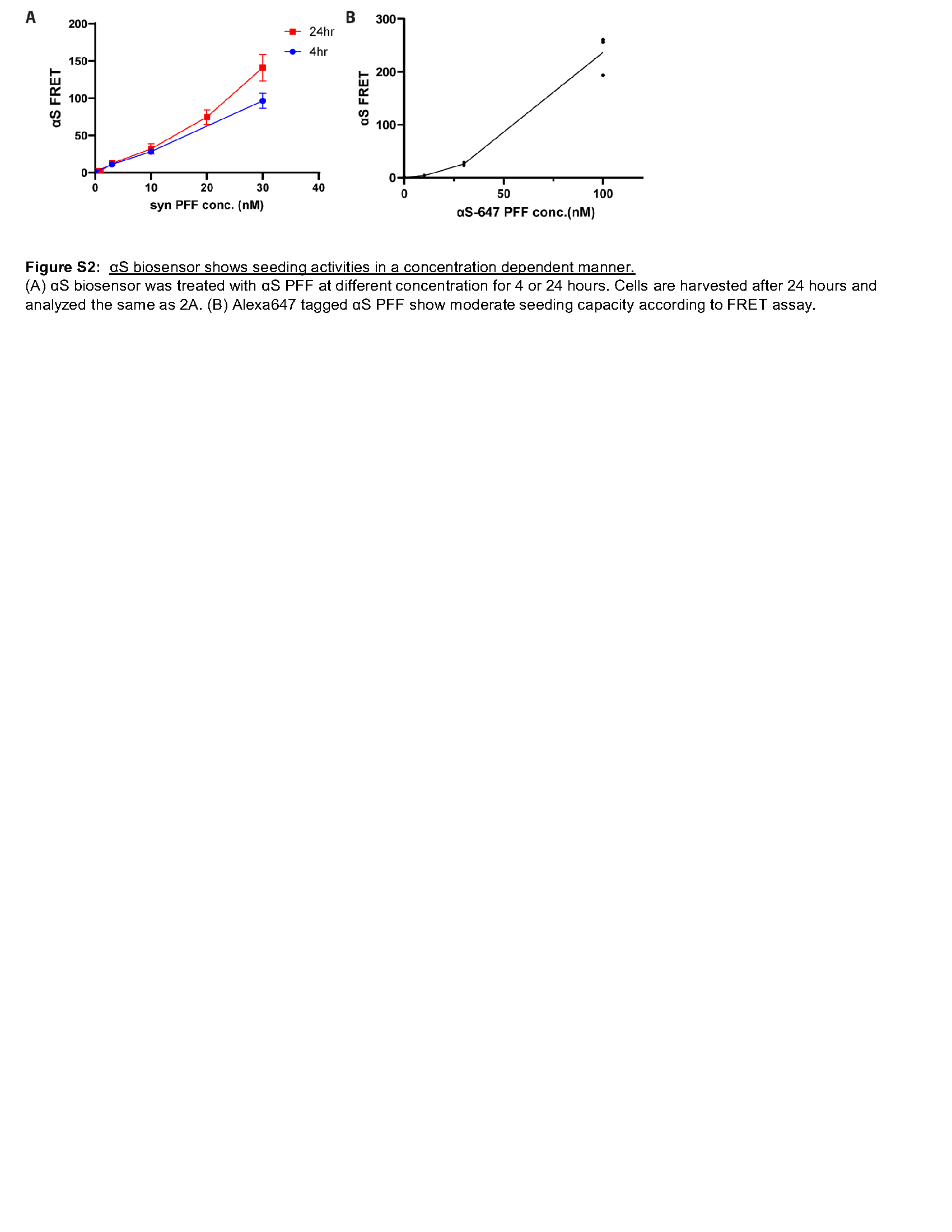
In order to probe the role of VCP specifically at the time of endocytic entry into the cytosol, we modified our seeding protocol to a four-hour application with lipofectamine and subsequent washout of αS PFF from the media and determined this to be sufficient to seed αS aggregation as measured by FRET in a concentration-dependent manner at 24 hours (Figure S2A). Consistent with αS PFF entry through the endolysosomal pathway, treatment of αS biosensors with Dynogo-4a, a dynamin I/II inhibitor with αS PFF, decreased FRET. Whereas a four-hour treatment at the time of αS PFF application with the lysosomal permeabilizing agent L-leucyl-L-leucine methyl ester (LLoMe) significantly increased FRET (Fig. 2B). Treatment of αS biosensors with the VCP inhibitor NMS-873 for four hours at the time of αS PFF application similarly increased seeding efficiency as measured by FRET (Fig. 2B). The application of two additional VCP inhibitors, CB-5083 and Eeyarestatin I (Eer1), during αS PFF seeding also increased seeding efficiency measured by FRET (Fig. 2B). This effect was αS PFF dependent since the application of VCP inhibitors or LLoMe in the presence of αS monomer did not increase FRET (Fig. 2B). A similar experiment adding the polyphenol (−)-epigallocatechin gallate (EGCG), an anti-amyloid agent at the time of αS PFF application, resulted in a decrease in seeding efficiency (Fig. 2B). Finally, the application of the proteasome inhibitor bortezomib or autophagy modulators (Rapamycin and 3-methyladenine (3-MA)) did not affect seeding efficiency (Fig. 2B).
To see whether the increased seeding efficiency with VCP inhibition was due to an increase in αS PFF uptake, we employed a fluorescently conjugated αS PFF (αS-PFF 647). αS-PFF 647 retains seeding capacity in αS biosensors (Figure S2B). A four-hour αS biosensor treatment with αS-PFF 647 in the presence of LLoME or NMS-873 did not increase the amount of internalized αS PFF 647 (Fig. 2C-D). Uptake was also unchanged when comparing scrambled and VCP siRNA KD (Fig. 2E). In contrast, the application of Dynogo-4a significantly decreased αS PFF 647 uptake (Fig. 2F). In addition, the increased seeding efficiency with VCP chemical inhibition or VCP siRNA knockdown was not due to an increase in the steady-state levels of αS as determined by immunoblot and αS fluorescence intensity via flow cytometry (Fig. 2G-K). VCP inhibition and knockdown induce ER stress and activate the unfolded protein response (UPR) [33, 34]. Treatment of αS biosensors with the ER stress inducing agents dithiothreitol (DTT), thapsigargin, and tunicamycin for 24 hours following the application of αS PFF decreased FRET efficiency as compared to vehicle controls suggesting that the effect of VCP inhibition on seeding was ER stress independent (Fig. 2L).
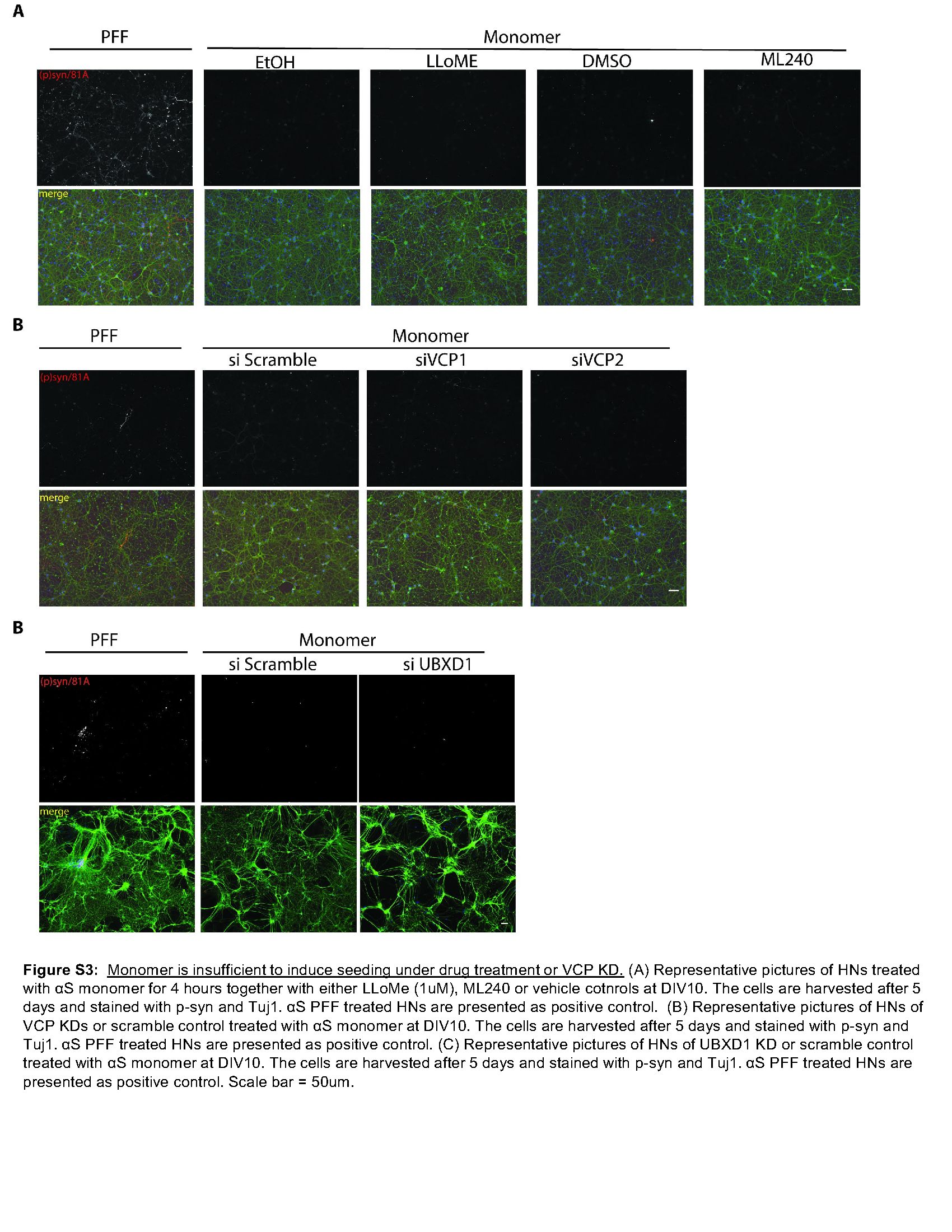
To explore the role of VCP in a more relevant system of αS seeding that does not require the use of the carrier Lipofectamine, we adapted a previously described assay that adds αS PFF to primary cultured hippocampal neurons (HNs) [35]. The addition of αS PFF to the media of HNs for four hours, followed by media exchange, resulted in detergent-insoluble, high molecular weight αS, and phospho-αS as measured by fractionation immunoblot or via immunofluorescence using a phospho-αS antibody in HNs after five days (Fig. 3A-B). The co-application of LLoMe and αS PFF for 4 hours followed by washout further increased the amount of phospho-αS as compared with the application of αS PFF and vehicle after five days (Fig. 3B-C). These results were αS PFF dependent since monomeric αS failed to generate phospho-αS staining (Figure S3A). We performed a similar assay and treated HNs for four hours with the reversible VCP inhibitor ML240 and αS PFF. Notably, the application of ML240 for four hours at the time of αS PFF application increased the level of phospho-αS as compared with vehicle-treated control, while drug-treated HNs with αS monomers show no phospho-αS (Fig. 3D-E, S3A). Treatment of primary HNs with two different shRNAs against VCP for four days before seed application further demonstrated an increase in an αS PFF-dependent increase in phospho-αS staining compared with scrambled shRNA control (Fig. 3F-G; S3B).
VCP disease mutation expression increases αS seeding.
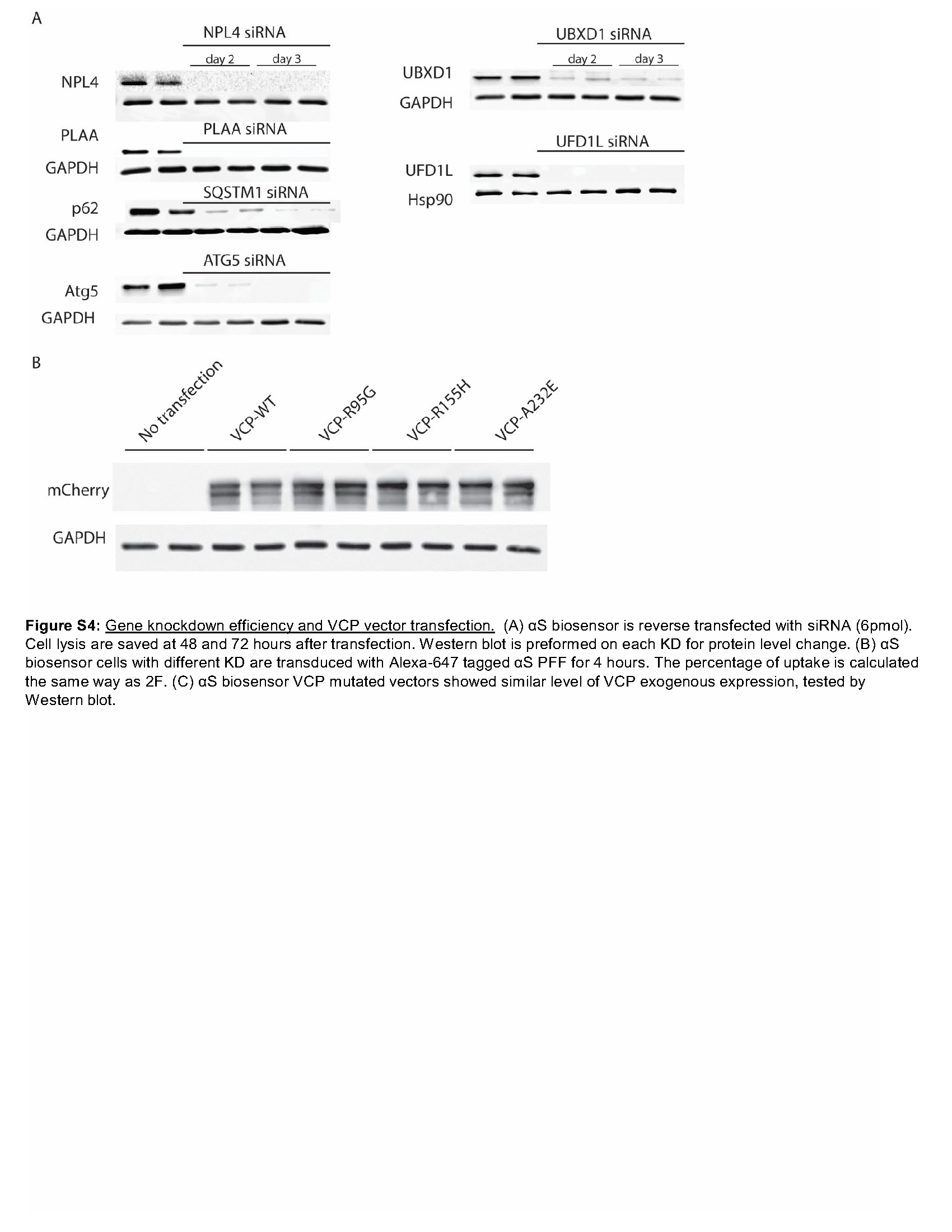
VCP disease mutations affect a subset of VCP dependent cellular processes such as endocytic trafficking, nutrient sensing, autophagosome maturation, and, more recently, lysophagy [7, 15]. This is due to an impairment in VCP mutant association with the adaptor UBXD1. We performed siRNA knockdown of VCP and the VCP adaptors UFD1, NPL4, UBXD1, and PLAA and the autophagy proteins ATG5 and SQSTM1 along with a scrambled control in αS biosensors. Following 48 hours of knockdown, αS biosensors were treated with αS PFF, and FRET was measured 24 hours later. Knockdown of VCP or its adaptor UBXD1 significantly increased FRET efficiency above controls (Fig. 4A and S4A). shRNA knockdown of UBXD1 in primary HNs 4 days prior to αS PFF application also increased phospho-αS staining as compared with scrambled shRNA control (Fig. 4B-C). To understand if VCP disease mutant expression increased αS seeding, similar to VCP and UBXD1 knockdown, we transfected αS biosensors with mCherry-tagged VCP-WT or one of three different VCP disease mutations (R95G, R155H, and A232E) for 24 hours. Treatment with αS PFF and quantitation of FRET efficiency 24 hours later in mCherry positive cells demonstrated an increase in FRET in VCP disease mutant expressing cells compared with VCP-WT control. Whereas cells not expressing mCherry did not show differences (Fig. 4D and S4C).
VCP-R155H mutation knockin mice have been previously generated and characterized [36]. We cultured primary HNs from VCPWT/WT and VCPRH/WT embryos, treated them with αS PFF, and then immunostained for phospho-αS five days later. VCPRH/WT had a significant increase in phospho-αS staining compared with VCPWT/WT controls (Fig. 4E-F).
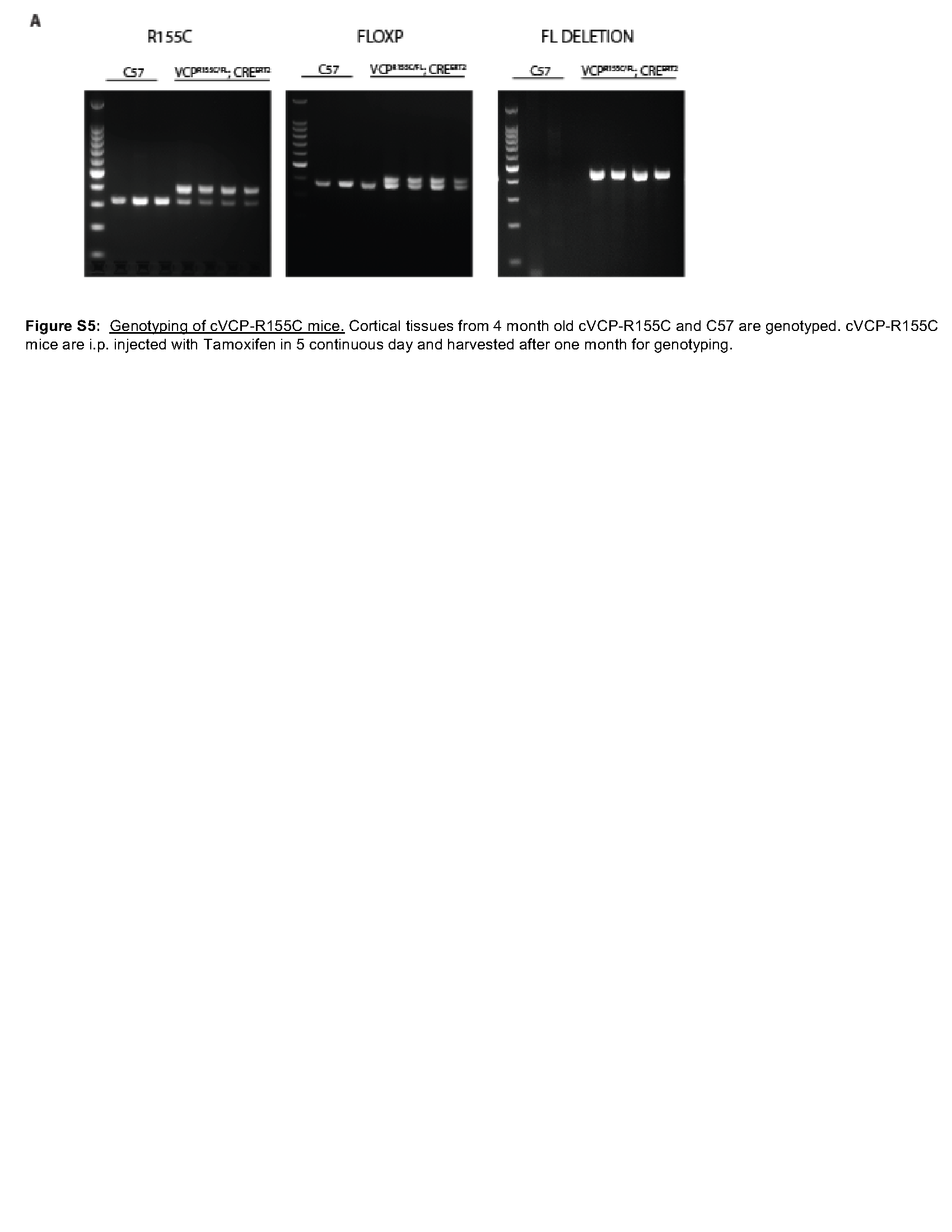
Using the same VCP-R155H mutation knockin mice, we examined the effect of pathogenic VCP mutations on αS seeding in vivo. VCPRH/WT mice display no neuronal loss, TDP-43 inclusions, or pathologic features consistent with autophago-lysosomal dysfunction up to 13 months old [37].To explore an additional VCP mouse model that only expresses a VCP disease mutant allele, we also used a mouse line that deletes the VCP-WT allele and only allows expression of a single VCP-R155C mutant allele following tamoxifen treatment (VCPRC/FL; Rosa26-Cre ERT2 (cVCP-RC) (Figure S5) [37]. Similar to our previous study, lysates from the cortex of VCPRH/WT have no changes in the levels of autophagic (LC3, Sequestosome-1/p62), ER stress (BiP/GRP78), or ubiquitinated proteins (Fig. 5A-B). Following five days i.p. tamoxifen treatment, cortical lysates from cVCP-RC mice have a 41% reduction in total VCP protein level but no changes in autophagic levels (LC3, Sequestosome-1/p62), ER stress (BIP/GRP78), or ubiquitinated proteins (Fig. 5A-B). However, high molecular weight ubiquitinated proteins, and SQSTM1 levels increase with age, as demonstrated by immunoblot of cortical lysates at 6 months, supporting that VCP dysfunction is present (Fig. 5C-D). We have previously demonstrated that an increase in Gal3 levels occurs prior to autophagic dysfunction in VCPRH/WT mouse muscle [17]. Similar to skeletal muscle, Gal3 and LAMP1 levels are increased in both VCPRH/WT and cVCP-RC mouse cortical lysates suggesting an accumulation of damaged late endosomes (Fig. 5A-B) [15, 17].
We injected 5ug αS PFF or PBS into the striatum of 4-month-old C57 control, VCPRH/WT, or cVCP-RC mice and harvested the brain after 3 months (Fig. 6A). Untreated control, VCPRH/WT, cVCP-RC mice, or mice treated with PBS had no phospho-αS staining in any brain regions (Fig. 6A-E). In contrast, C57 control mice injected with αS PFF had a significant increase of phospho-αS in multiple brain regions (Fig. 6A-E). This increase was significantly increased above that of injected C57 mice in the anterior and posterior cortices of VCPRH/WT, and cVCP-RC injected with αS PFF (Fig. 6A-E). Other brain regions such as the amygdala and substantia nigra trended toward an increase in phospho-αS staining but did not reach statistical significance (Fig. 6F-G).
VCP disease mutations enhance TDP-43 seeding.
A subset of VCP patients has Parkinsonism and post-mortem evidence of αS pathology. However, most patients have TDP-43 inclusions in the CNS and muscle [8–10]. To evaluate the role of VCP in the seeding of TDP-43, we developed a TDP-43 seeding assay in primary hippocampal neurons. The addition of TDP PFF to HNs resulted in the appearance of phosphorylated TDP-43 Ser409/410 (pTDP) positive puncta in a concentration-dependent manner (Fig. 7A-B) and redistribution of pTDP from the nucleus to the cytoplasm (Fig. 7C) after 5 days. pTDP staining was not increased or altered when HNs were treated with αS PFFs (Figure S6). Fractionation of lysates from HNs one or 5-day post-treatment with buffer, non-aggregated TDP-43 (monomeric TDP-43), or TDP-43 aggregated at room temperature for 24 hours (TDP-43 PFF) and subsequent immunoblot for pTDP revealed an increase in high molecular weight TDP-43 in the RIPA insoluble fraction of TDP PFF treated HNs (Fig. 7D). In addition, TDP-43 PFF induced cytosolic pTDP-43 puncta that co-localized with Sequestosome-1 and TIA1 similar to pathologic TDP-43 inclusions in patients (Fig. 7E). As seen with VCP mutant expression in TDP-43 biosensors, treatment of primary HNs from VCPWT/WT and VCPRH/WT embryos with TDP-43 PFF revealed an increase pTDP-43 puncta in VCPRH/WT HNs compared with VCPWT/WT HNs (Fig. 7F-G).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}