Materials
The human neuroblastoma cell line SK-N-MC was obtained by Korean Cell Line Bank (Seoul, Korea). Fetal bovine serum (FBS) was purchased from Hyclone (Logan, UT, USA). Antibiotics and serum replacement (SR) were purchased from Gibco (Grand Island, NY, USA) The antibodies of β-actin, cat-PKA , CREB, p-CREB (Ser113), p-CaMKII (Thr 286), CaMKII, CaM, p-JNK, ASC, BNIP3, NIX were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The antibodies of Drp1, p-Drp1 (Ser 616), PINK1, Cleaved caspase-3 and Small interfering RNAs (siRNAs) for JNK1 were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). The antibodies of TOMM20, parkin were purchased from Abcam (Cambridge, England). The antibody of NR2B was purchased from Invitrogen Corporation (Camarillo, CA, USA). The antibody of α-tubulin purchased from Sigma Chemical Company (St. Louis, MO, USA). The antibodies of COX4, JNK1 (MAPK8) were purchased from CusaBio (Houston, TX, USA). The antibody of NLRP3 was purchased from AdipoGen Life Sciences. The antibodies of caspase-1, LC3 purchased from Novus Biologicals (Littleton, CO, USA). CM-H2DCFDA, MitoSOX™ Red, Mitotracker™ Green, Mitotracker™ Red were obtained from Thermo Fisher (Waltham, MA, USA). The 14–22 amide was obtained from Calbiochem (Merck Millipore). NAC, MitoTEMPO, SP600125, Ac-YVAD-cmk, Mdivi-1, KN-93, MK-801 were purchased from Sigma Chemical Company (St. Louis, MO, USA). Small interfering RNAs (siRNAs) for CREB1, CASP1 and non-targeting (NT) were purchased from Dharmacon (Lafayette, CO, USA).
Cell culture
The SK-N-MC cells were cultured in high-glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS and 1% antibiotics. Cells were seeded in 60 or 100 mm diameter culture dishes, or in 6- or 12-well plates and incubated at 37°C incubator with 5% CO2. When cells were grown 60-70% confluence, the medium was exchanged with serum-free medium containing 2% SR prior to experiments.
Real time quantitative PCR
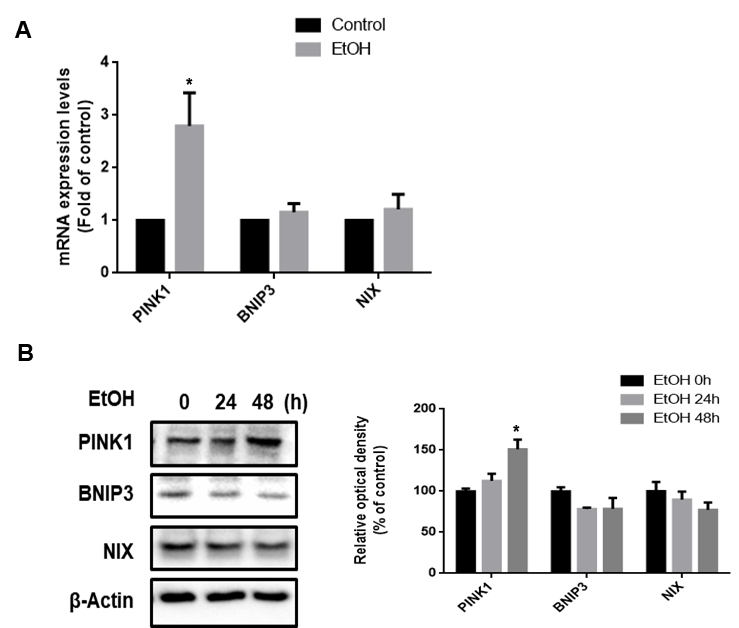
RNA was extracted from SK-N-MC using MiniBEST Universal RNA Extraction Kit (TaKaRa, Otsu, Shinga, Japan). Reverse transcription polymerase chain reaction (RT‐PCR) was carried out using 1 μg of extracted RNA and a Maxime™ RT‐PCR premix kit (iNtRON Biotechnology, Sungnam, Korea). RT‐PCR was performed for 60 min at 45°C to cDNA synthesis and 5 min RTase inactivation at 95°C. The cDNA was amplified using Quanti NOVA SYBR Green PCR Kits (Qiagen, Hilden, Germany). Real-time quantification of RNA targets was carried out using RotorGene 6000 realtime thermal cycling system (Corbett Research, NSW, Australia) with mRNA primers and 1 μg of cDNA sample. Human primer sequences are described in Table 1. The Real-Time PCR was performed as follows: 15 min at 95 °C for DNA polymerase activation; 15 s at 95 °C for denaturing; and 40 cycles of 15 s at 94 °C, 30 s at 56 °C, and 30 s at 72 °C. Data were collected during the extension step (30 s at 72 °C), and analysis was performed with software provided by Rotor‐Gene 6000 Series software (Qiagen, Hilden, Germany) to verify the specificity and identity of the PCR products.
Western blot analysis
Cells were collected by using scraper after being washed once with cold PBS and incubated for 30 min on ice with RIPA buffer (ATTO Corporation, Tokyo, Japan) and a proteinase and phosphatase inhibitor (Thermo Fisher). The lysate were then cleared by centrifugation (15,000 rpm, 4°C, 20 min). The Protein concentration was determined by BCA assay kit (Bio-Rad, Hercules, CA, USA). Samples containing 10 ug of protein were prepared for 6 - 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked with 5% skim milk (Gibco) for 50 min and blocked membrane was washed with TBST solution 4 times every 8 min. After that, membrane was incubated with primary antibody overnight at 4 °C. The membrane was washed and incubated with HRP-conjugated secondary antibody (1:10 000) at room temperature for 2h. The western blotting bands were visualized by using chemiluminescence (BioRad, Hercules, CA, USA). Densitometric analysis was performed with the Image J software (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD, USA).
Measurement of calcium
Fluo 3-AM was used to measure intracellular calcium levels. The cells on 6-well dishes washed with a PBS once and then incubated in PBS containing 2 μM Fluo 3-AM for 30 min at 37 °C in dark. Cells were treated with a 0.05% trypsin for 3 min and then centrifuged at 1,500 g for 5 min. After centrifugation, cells were washed once with PBS, followed by suspending the cells in 400 μL PBS. Relative fluorescence intensity (RFI) of Fluo 3-AM was measured using flow cytometry (CytoFlex; Beckman Coulter, Fullerton, CA, USA).
Measurement of intracellular reactive oxygen species levels
The cells were plated on 6- or 12-well dishes. Cells were washed once with PBS and incubated with 1 μM CM-H2DCFDA for 30 min at 37°C in dark. Cells were treated with a 0.05% trypsin for 3 min and then centrifuged at 1,500 g for 5 min. Next, cells were washed once with PBS, followed by suspending the cells in 400 μL PBS. DCFDA staining was detected via flow cytometry (CytoFlex; Beckman Coulter, Fullerton, CA, USA).
Measurement of mitochondrial ROS generation
The measurement of mitochondrial ROS generation was performed by using MitoSOX™ Red staining. Cells were washed once with PBS and incubated with 10 μM MitoSOX™ for 15 min at 37°C in dark. Cells were then treated with a 0.05% trypsin for 3 min and then centrifuged at 1,500 g for 5 min. Collected cells were suspended in 400 μL PBS. MitoSOX™‐positive cells were detected by using flow cytometry (Beckman Coulter).
Measurement of mitochondrial membrane potential and mitochondrial volume
To evaluate the mitochondrial membrane potential and volume, a TMRE (Sigma-Aldrich) and Mitotracker™ Green staining were used, respectively. After treatment, cells were incubated in 50 nM TMRE or 200 nM of Mitotracker™ for 20 min at 37°C in dark. Cells were then treated with a 0.05% trypsin for 3 min and then centrifuged at 1,500 g for 5 min. Collected cells were suspended in 400 μL PBS. Fluorescence intensities of TMRE or Mitotracker™ Green were detected by using flow cytometry.
Annexin V/PI apoptosis detection
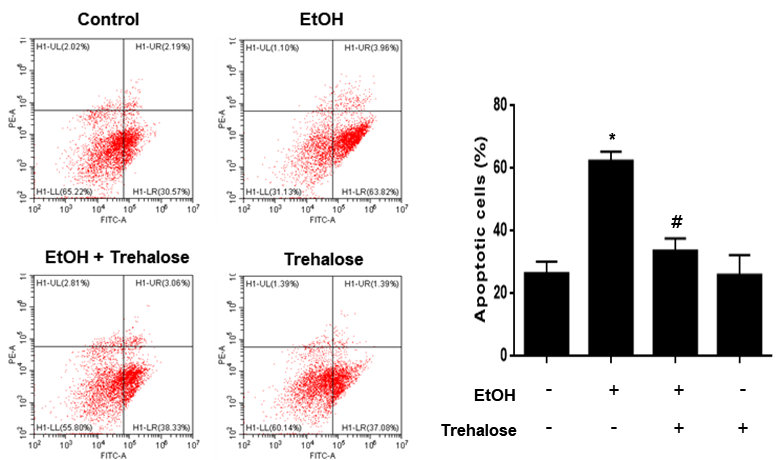
To measure apoptosis of cells, Annexin V and PI double staining was performed by using an annexin V/PI apoptosis detection kit (BD Bioscience, Franklin Lakes, NJ, USA) according to the supplier's manual. Cells were detached with 0.05% trypsin and counted 1×105 cells. Cells were then centrifuged at 1,500 g for 5 min. Collected cells were suspended in the binding buffer supplied by the kit, and immunostained with AnnexinV-FITC (5 μL) and PI (5 μL) for 20 min at room temperature in dark. Cell apoptosis was measured by using flow cytometry (Beckman Coulter). Data were analyzed by using CytExpert software (Beckman Coulter). AnnexinV-positive and PI-negative (Q4), AnnexinV-positive and PI-positive (Q2), and AnnexinV-negative and PI-positive (Q1) were considered as early apoptotic, late apoptotic and necrotic cells, respectively. AnnexinV-negative and PI-negative (Q3) cells were considered viable. To measure the percentage of total apoptotic cells, the following formula was used: Apoptotic cells = Q2 + Q4.
Immunocytochemistry
Cells were cultured on a confocal dish and fixed with 4% paraformaldehyde (Sigma Aldrich) for 10 min. 0.1% Triton X-100 was used for permeabilization for 5 min. To inhibit nonspecific binding of antibodies, cells were incubated with 1% normal goat serum for 30 min. Next cells were incubated with 1:100 dilution of primary antibody for 2 hours at room temperature and washed with PBS three times. After washing, cells were incubated with Alexa Fluor 488 or 555-conjugated secondary antibody (1:300) in dark for 1 h in room temperature. Stained images were visualized by a super-resolution radial fluctuations (SRRF) imaging system (Andor Technology, Belfast, UK). Relative Fluorescence intensity was analyzed by using ImageJ software.
Mitochondria morphology
The cells were plated on confocal dish and incubated with Mitotracker™ Red (200 nM) for 30 min at 37 °C. To visualize Mitotracker™ Red-stained cells, we used a super-resolution radial fluctuations (SRRF) imaging system. Analysis of images was performed by using FIJI software. Form factor (FF = perimeter2/4π * area) and aspect ratio (AR) were used for analysis of mitochondrial fragmentation. As both FF and AR approaches 1, they represent a circular shape, but the both parameters increase, the mitochondria morphology becomes elongated.
Co-immunoprecipitation
Cells were lysed with the co-immunoprecipitation buffer (1% Triton X-100 in 50 mM Tris-HCl [pH 7.4] containing 150 mM NaCl, 5 mM EDTA, 2 mM NA3VO4, 2.5 mM Na4PO7, 100 mM NaF, 200 nM microcystin lysin-arginine, and protease inhibitor). Primary antibodies were immobilized with SureBeads™ Protein G magnetic beads (BioRad, Hercules, CA, USA, #161-4021). Immobilized magnetic beads were washed three times with PBST and then incubated with cell lysates (350 μg) for 12 h at 4 ℃. Beads were washed three times with PBST and incubated with elution buffer (20 mM glycine pH 2.0) for 5 min. 1 M phosphate buffer and sample buffer were added to the samples.
Small interfering RNA (siRNA) transfection
Prior to ethanol treatment, cells were incubated with 25 nM of the indicated siRNAs and transfection reagent TurboFect™ (Thermo Fisher, Waltham, MA, USA, #R0531) for 24 h in serum-free medium containing 2% SR. The non-targeting (NT) siRNA was used as the negative control.
Mitochondria fraction
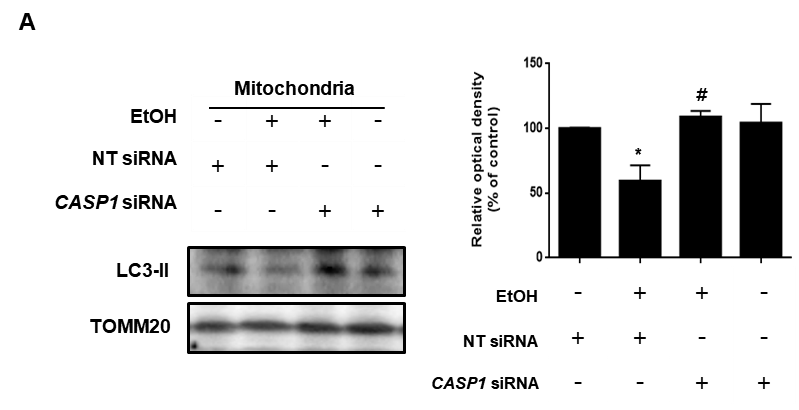
Mitochondrial fraction was performed using mitochondria isolation kit according to the manufacturer's instructions (Thermo Fisher Scientific). Cytoplasmic and mitochondrial proteins were extracted from cells according to the manufacturer's instructions. Briefly, collected cells were incubated in Reagent A for 2 min on ice and then cell lysate was incubated with Reagent B for 5 min. Next, Reagent C was added to the cell lysate. After centrifugation, supernatant was used as a cytosolic fraction. The pellet was lysed with 2% CHAPS in Tris-buffered saline (25 mM Tris, 0.1 M NaCl, pH 7.2) solution and used as a mitochondrial fraction after centrifugation.
Trypan blue exclusion cell viability assay
Cells were incubated with a 0.05% Trypsin to detach the cells. To identify dead cells, 0.4% trypan blue was added to the cell. Stained (dead) and unstained (live) cells were counted by using a Petroff–Hausser counting chamber (Hausser Scientific, Horsham, PA, USA). Cell viability = [{1− (number of trypan blue‐stained cells/number of total cells)} × 100].
Statistical analysis
Results are expressed as mean ± standard error of mean (S.E.M). Differential among experimental groups were analyzed by analysis of variance (ANOVA), and two group analysis was conducted by using Student's t test. P value of < 0.05 was considered statistically significant.
{kind=link}
{kind=link}
{kind=link}