Since the living organisms continuously suffer from many stresses such as reactive oxygen species produced by energy reaction, ionizing radiation, ultraviolet radiation, toxic chemical agents, and other environmental factors during normal activities[32], the mechanism that protects cells from various stress remains incompletely understood. These damages may impact nucleic acids, lipids, and proteins and lead to mutations or genome aberrations that result in genetic diseases, developmental defects, cancer, and even death[33]. To handle these serious damages, cells had to develop an intricate network signaling pathway to deal with various stress[34]. Maintaining genomic stability is also especially important for the tumor cells. Losing of one or more DDR pathways has been proved to be part of the stage of cancer development. Whereas, once defects in DDR may lead to more dependence on the remaining pathways[35]. As an important phase Ⅱ detoxifying enzyme, GSTpi was detected with high expression in many tumors and cancer cell lines[5]. Nevertheless, the high expression level of GSTpi was found to be related to chemoresistance, and studies on this mechanism mainly focused on the detoxification and antioxidant functions of GSTpi[36, 37]. However, most anticancer drugs are not substrates of GSTpi, the reasons for chemoresistance remain unclear. Recently, Dong XL et al. have reported that GSTpi enhanced the resistance of breast cancer to ADR through autophagy[10], which is independent of its enzyme activity. Consistently, the previous mechanisms involving the non-transferase activity of GSTpi that participates in DNA damage remain unclear[38]. The functional repertoire of GSTpi has broadened significantly beyond its original transferase activity in many biological processes. Our investigation into how GSTpi protects cells from DNA damage provides a physiologically relevant context to understand the reason of the high expression of GSTpi in breast cancer cells.
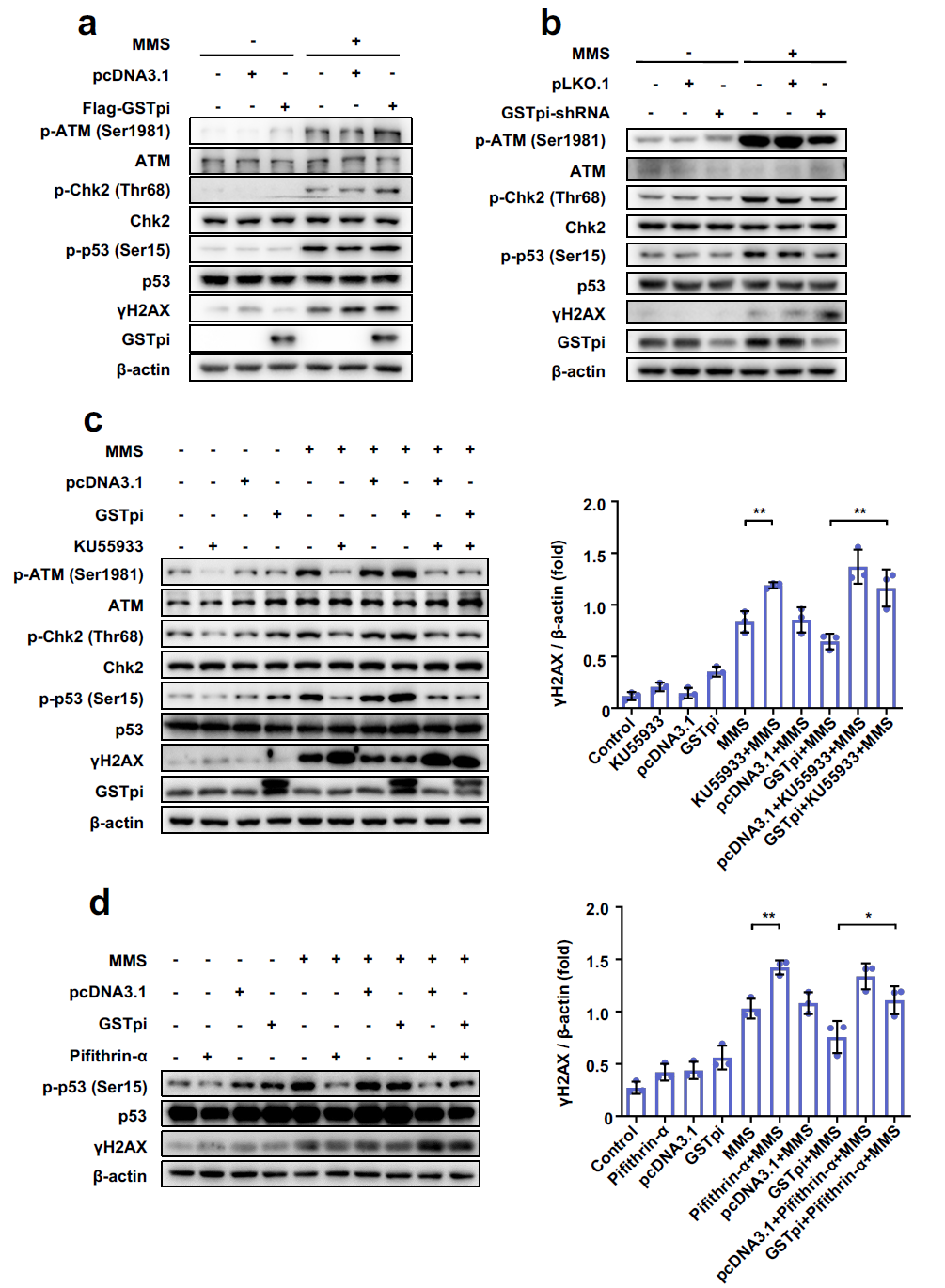
Based on some previous studies on protecting cells upon the UVC irradiation or MMS treatment[39, 40], we initiated our research to investigate the influence of GSTpi on MMS triggered DNA damage. Our finding revealed that the level of γH2AX was decreased by GSTpi after MMS stimulation. Moreover, both immunofluorescence assay and single-cell gel electrophoresis demonstrated that GSTpi has a vital effect on DDR. Earlier studies have reported that the nuclear GSTpi protects DNA against damage[11, 19]. Recently, Zhou Y et al. found that GSTpi enters the nuclear and interacts with HMGB1 in response to LPS[24]. Consequently, we conjectured that the capacity of GSTpi to improve DNA damage repair may contribute to its nucleus translocation. Interestingly, our results showed that GSTpi entered the nucleus when DNA damage occurred. This discovery enriches the function of the nuclear GSTpi, which breaks the limitation of previous research on the cytosolic GSTpi[41, 42].
When DNA damage occurred, some specific sensor proteins including ATM, ATR, and DNA-PKcs recruit to damage sites and initiate a DDR[43, 44]. ATM and ATR have similar sequences to lipid kinases of the PI3K family[45], they can selectively phosphorylate and activate the serine-threonine checkpoint effector kinases which are called Chk2 and Chk1[46]. Our results demonstrated that GSTpi activated the ATM-Chk2 pathway against MMS-induced DNA damage. Some known substrates of Chk2 including p53, MDMX, Cdc25, and BRCA1[46–49]. Importantly, we found overexpression of GSTpi enhanced the phosphorylated p53, it can also induce the cell cycle arrest in the G2/M phase and gain more time for cells to proceed DNA damage repair.
It has been reported that the MRN complex can not only sense the damage sites but also activate ATM[28, 50]. Particularly, the role of NBS1 in the recruitment of ATM to DNA damage sites is of vital importance[51], so we next explored the relationship between NBS1 and GSTpi. Interestingly, we found for the first time that phosphorylated GSTpi can promote itself and NBS1 into nuclear during DNA damage. What’s more, the apoptotic rate of breast cancer cells was significantly increased when ADR was combined with GSTpi phosphorylated inhibitor and autophagy inhibitor in comparison to treatment with ADR alone; however, NBS1 nuclear translocation could only inhibit by GSTpi phosphorylated inhibitor. These findings displayed a new function of phosphorylated GSTpi that protects breast cancer cells against anti-tumor drugs and resulted in the drug resistance through two individual pathways including autophagy and NBS1 nuclear translocation.
Protein post-translational modification (PTM) is closely related to the activity, stability, localization, interactions, or folding of proteins[52], implying that one or more PTMs to NBS1 may cause its change in protein expression and its translocation to the nucleus. Then we focused on the post-translational modification to NBS1 mediated by GSTpi. It seemed that the increased ubiquitination degradation of NBS1 was induced by GSTpi without MMS treatment. On the contrary, GSTpi inhibited the ubiquitination degradation of NBS1 when upon MMS. We further investigated the combination among GSTpi, NBS1, and the ubiquitin-protein ligase (E3 ligase). Our finding showed that GSTpi enhanced the interaction of NBS1 and Skp2 in unstimulated cells, conversely, when DNA damage occurred the interaction weakened, which indicated that GSTpi can protect NBS1 from ubiquitination degradation mediated by Skp2 when DNA damage occurred. Based on the above results, we provide another novel mechanism of the post-translational modification of NBS1 that participates in sensing DNA damage sites. We also speculated Skp2 may trigger K48-linked ubiquitination of NBS1 and the process regulated by GSTpi. However, given the capacity of multiple PTMs to regulate NBS1, we cannot completely conclude that the K48-linked ubiquitination of NBS1 contributes to the activation of ATM kinases either independently or in combination with other modifications. However, these speculations will be verified in future experiments.
In summary, our present study demonstrated that when DNA damage occurred, GSTpi can be phosphorylated at Ser184, and the phosphorylated GSTpi inhibited the ubiquitination degradation of NBS1 mediated by Skp2 in the cytoplasm, then combined with NBS1 and entered nuclear more to trigger the G2/M checkpoint arrest through activating the ATM-Chk2-p53 signaling pathway (Fig. 7a). Based on our findings, we explained a novel mechanism of GSTpi protecting cells against DNA damage. This new molecular insight into the new function of GSTpi will offer us a novel strategy for solving the problem of breast cancer drug resistance.
{kind=link}