Synthesis and crystal structure. Dy-I and Y-I were synthesized using the Krätschmer-Huffman arc-discharge method24. Graphite rods, filled with MNi2 alloy (M = Dy and Y) and melamine powder as solid nitrogen source34, were evaporated under helium atmosphere at high temperatures, resulting in the carbon-based solid. Multi-stage high performance liquid chromatography (HPLC) was then applied to isolate pure Dy-I and Y-I from the extract of raw soot (see Supplementary Information for detailed information). To determine the molecular structure through single crystal XRD analysis, a black crystal was obtained by co-crystalizing Dy-I with NiII(OEP) molecule (OEP is the dianion of octaethyl porphyrin).
The crystal system of the obtained Dy-I·NiII(OEP)·C6H6 co-crystal falls into the monoclinic C2/m space group, where the asymmetric unit cell contains one half of the NiII(OEP) molecule and two halves of the C3v(8)-C82 cage, regardless of the nitrogen substitution. Note that due to the similar electron density of N and C, direct identification of the exact location of the N atom on the cage is not feasible and will be discussed later. Within the fullerene cage, two fully ordered N atoms are assigned. The other two N are generated by symmetric operation, leading to the unequivocal determination of a planar cyclo-N4 ring (Figure 1a). The averaged N-N distance is determined as 1.609(9) Å, which is longer than those of reported polymeric nitrogen compounds with double or aromatic bonds10,11, as well as conventional N-N single bond in hydrazine (1.45 Å)35. However, it is similar to the values of crystalline N2O4 (1.64 Å)1 and metal-triazane cation Ag2(N3H5)32+ (1.6(1) Å)36, suggesting a weak bonding character between each two adjacent N atoms in the cyclo-N4 unit. This is likely due to the ring strain and metal-ring interaction that weaken N-N bonds, in retrospect to the longer bond length in the silver ion-complexed cyclo-N5- than that of the pristine metal-free anion13.
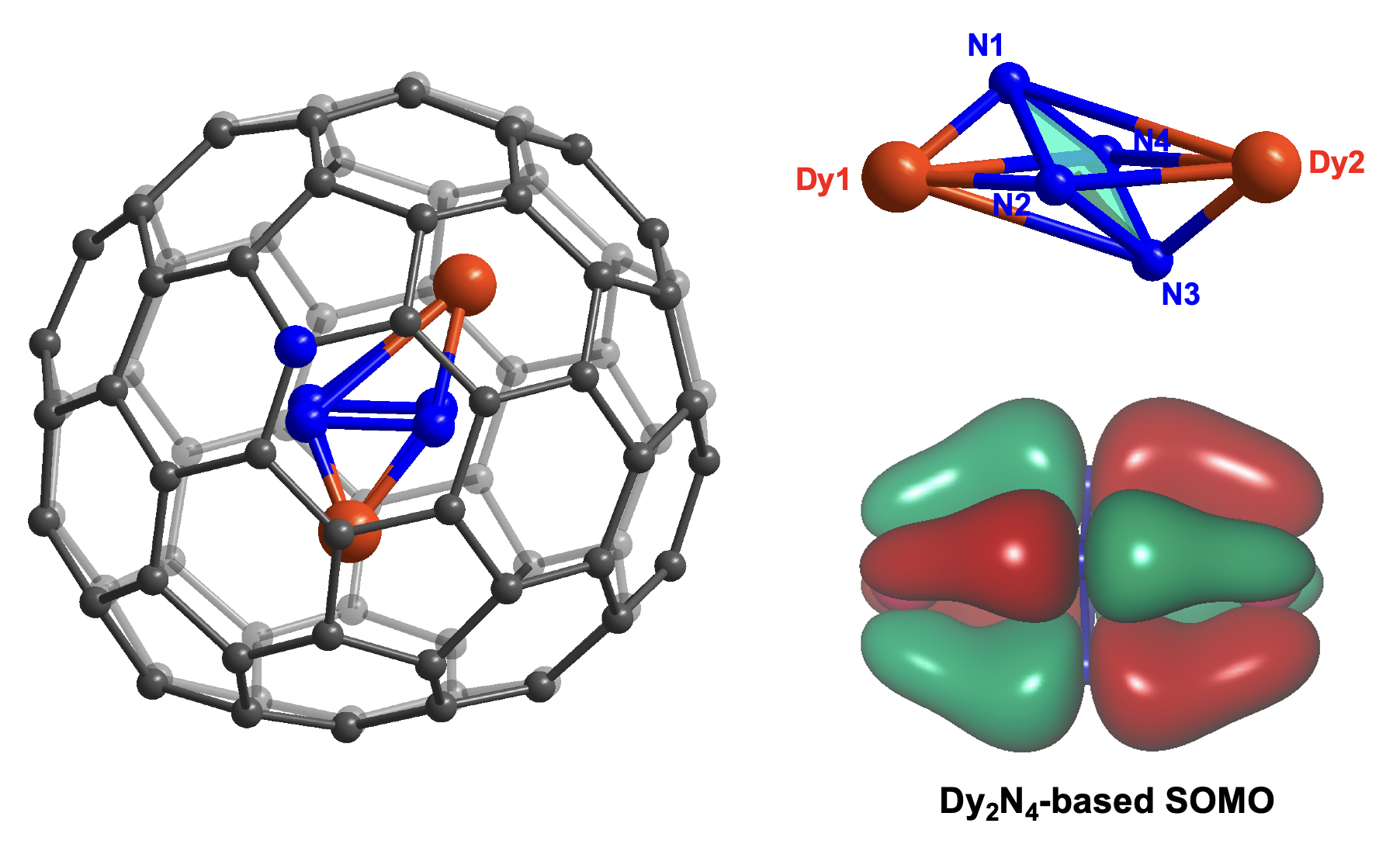
On the other hand, two metals are highly disordered. A total of twenty Dy sites with occupancy varying from 0.03 to 0.27 are widely distributed on two sides of the cyclo-N4 plane. The latter stays in the center of the cage and is almost parallel (dihedral angle: 176.73˚) to the NiII(OEP) molecule (Figure 1b). The major sites Dy1 and Dy2, with a Dy-Dy distance of 3.702(5) Å, unexpectedly reside at off-center positions with respect to cyclo-N4, featuring a distorted octahedron with inverse-sandwich Dy-N4-Dy coordination (Figure 1c). This results in a wide range of Dy-N distances from 1.527 Å to 2.828 Å. Despite the fact that such short Dy-N distances are rarely seen in any organometallic complex, it is nevertheless not uncommon for endohedral metallofullerene compounds in which special chemical bond can be found due to severe disorder of metal atoms and confined inner space of fullerene cage. For instance, the C-C distances of M2C2 (M = La, Er and Lu)37-39 clusters in carbide clusterfullerenes are only about 0.9 Å, much shorter than the average length (1.2 Å) of a CºC triple bond. In addition, Dy1 and Dy2 are located below two hexagon carbon rings, showing a quasi-η6 metal-cage coordination mode (Figure S3.2). The potential energy surface for metal inside the cage may also be influenced by the existence of porphyrin in the co-crystal.
Computational studies. In order to verify the peculiar structure of Dy-I and probe in particular the bonding nature of the cyclo-N4 unit, density functional theory (DFT) calculations were performed starting from the crystal structure. The resulted molecular structure of Dy-I is shown in Figure 2a. In line with the crystal structure, the planar cyclo-N4 ring is maintained, while they differ in the fact that the calculated cyclo-N4 unit symmetrically complexes to two dysprosium ions. The Dy-N distances fall in the range of 2.28-2.31 Å, rendering an ideally octahedral Dy-N4-Dy configuration. As discussed above, the crystal structure deviates from this ideal gas phase model as a result of the complicated circumstance in the Dy-I·NiII(OEP)·C6H6 co-crystal. Concerning the nitrogen location on the fullerene cage, the hexagon/hexagon/pentagon (665) junction substituted structure (Figure 2a) is energetically favorable based on the previous studies of M2@C79N (M = Y and Gd)28,40; besides, nitrogen tends to reside in the middle of metal ions, thus avoiding short metal-nitrogen distances in La3N@C79N41 and Y2@C81N42. This is also confirmed by the calculations of Y-I, where Dy3+ is replaced by diamagnetic Y3+ with similar ion radius (Figure S4.1). Notably, the configuration of the encapsulated cluster is not affected by the nitrogen locations on the cage.
The bonding nature of Dy-I is next illustrated by electron localization function (ELF) mapping on the cyclo-N4 plane. Figure 2b shows a salient electron-density accumulation between each nitrogen pair, validating four N-N bonds in cyclo-N4. On the other hand, nitrogen lone pairs are assigned to areas around four nitrogen atoms with relatively diffuse electron localization, which contributes to the strain relief of the tetranitrogen ring43. Frontier molecular orbital (MO) analysis of this open-shell compound reveals a cluster-based singly-occupied MO (SOMO) (Figure 2c). As a consequence, spin density is also distributed within the inverse-sandwich Dy-N4-Dy cluster and shares the same shape with SOMO (Figure 2d), indicating efficient one-electron charge transfer from C81N to Dy2N4, which is similar to the circumstances in M2@C79N (M = Y and Gd)28,40. In this sense, the fullerene cage stabilizes the enclosed radical, giving rise to moderate SOMO-LUMO gaps of Dy-I (1.26 eV) and Y-I (1.30 eV) (Figure 2e).
As the entrapped six-atom cluster is of significant interest to us, its multi-center bonding motifs in Y-I were further elucidated by partially localized orbital analysis using adaptive natural density partitioning (AdNDP) method44. Among the cluster-based AdNDP orbitals, Figure 3a-c show four N-N single bonds, two Y-N dative bonds and one N-N π bond, respectively; Figure 3d presents one half-occupied π* anti-bonding orbital perpendicular to the tetratomic nitrogen ring, which renders a SOMO-like distribution with substantial Y(4d) contribution from two metals (a 6c-1e bond). To sum up, 15 p-type electrons have been clearly allocated to the cyclo-N4 ring, yielding a cyclo-N43- radical anion and a 3+ formal charge of the Y2N4 unit, while C81N adopts a 3- ionic model and thus is isoelectronic to the stable C3v(8)-C824- anion.
Spectroscopic and electrochemical properties. The composition of Dy-I and Y-I were confirmed by laser-desorption ionization time-of-flight (LDI-TOF) mass spectrum, showing well-matched theoretical and experimental isotopic distributions around the prominent molecular ion peaks accordingly with mass-to-charge ratio of m/z = 1367.8 and 1219.7 (Figure 4a and Figure S2.2). Their visible-near-infrared (vis-NIR) absorption spectra (Figure 4b) both feature two characteristic peaks at wavelength of 720 and 910 nm, which resemble those of C3v(8)-C82 based metallofullerenes hosting metal carbide (Sc2C245 and Dy2C2) and dimetallic (Sc246 and Er247) clusters. This further verifies that Dy-I and Y-I possess an identical molecular structure with an azafullerene C81N cage derived from one nitrogen substitution of C3v(8)-C82.
The EPR spectrum of Y-I solution at room temperature shows rich hyperfine patterns from 89Y (I = 1/2) and 14N (I = 1) nuclei with significant anisotropy. The hyperfine tensor components A^ and A|| of two inequivalent 89Y centers (Y1/Y2) are 208/237 MHz and 213/242 MHz, respectively; four 14N nuclei exhibit an equivalent hyperfine tensor with A^(14N) = 13.0 MHz and A||(14N) = 19.5 MHz; the g-tensor components are determined as g^ = 1.9780 and g|| = 1.9775. The large A(89Y) values and g factor below 2 indicate that significant spin density is localized on the two Y atoms, which is consistent with the spatial distribution of SOMO fully enclosed inside the cage and is comparable to the single-electron Y-Y bond in Y2@C79N and Y2@C80-CH2Ph32,48. The 14N hyperfine splitting is similar to the value of 16.3 MHz (5.8 G) for N23--radical-bridged diyttrium complex49. In contrast to the SOMO of the latter which is mainly localized at its N2 moiety, the highly diffuse radical character of Y-I also suggests noticeable metal-N4 interaction, which would weaken N-N bonds whereas contribute to the stabilization of this special six-atom cluster inside the fullerene cage.
The redox behavior of Dy-I was investigated in an ortho-dichlorobenzene (o-DCB) solution by means of cyclic voltammetry to display two reversible oxidation steps and four reversible reduction peaks (Figure 4d). Compared with the results of other C3v(8)-C824- based metallofullerenes (Table 1), the redox property of Dy-I is distinct. The most striking differences lie in the first oxidation (oxE1) and reduction (redE1) steps, in which the cluster-based SOMO of Dy-I is largely involved. In spite of their very different redox behaviors, the electrochemical gap of Dy-I (1.13 V) is still close to those of M2@C3v(8)-C82 (M = Sc and Er). A closer analysis (Figure S4.4) reveals that one-electron oxidation of Dy-I results in an expected transformation of N43- to N42-. On the contrary, the high-lying unoccupied cluster-based orbital precludes the reduction of N43- to N44-, where each N obeys octet rule to form four N-N single bonds. Instead, the cyclo-N43- unit donates the unpaired electron to the cage-based LUMO of Dy-I upon reduction, leading to a [Dy2]6+[N4]2-@[C81N]5- electronic configuration of Dy-I- anion. Noteworthy is that this eccentric electronic property of the N4 ring is in accordance with the fact that the N4R4+ radical cation is more stable than its neutral form in solution50, suggesting a unusual bonding tendency in cyclo-N4 which is likely attributed to the strain of the four-membered nitrogen ring.
Table 1. Redox potentials (V vs Fc/Fc+)a and electrochemical band gaps (ΔEgap)b of Dy-I and related metallofullerenes possessing a C3v(8)-C82 cage.
|
Compounds
|
oxE2
|
oxE1
|
redE1
|
redE2
|
redE3
|
redE4
|
ΔEgap
|
Ref.
|
|
Dy-I
|
0.85c
|
0.46c
|
-0.67c
|
-1.29d
|
-1.62c
|
-2.08d
|
1.13
|
this work
|
|
Er2@C3v(8)-C82
|
-
|
0.11
|
-1.09
|
-1.33
|
-1.76
|
-2.49
|
1.20
|
46
|
|
Sc2@C3v(8)-C82
|
-
|
0.02
|
-1.16
|
-1.53
|
-1.73
|
-2.02
|
1.18
|
47
|
aRedox potentials in V are measured vs. ferrocene couple. bΔEgap = oxE1 - redE1. cHalf-wave potential. dPeak potential.
{kind=link}