PARP16 is upregulated in HT22 cells after OGD/R and in brain of mice after MCAO
HT22 cells were cultured and injured by anaerobic bags to make OGD/R model. The proteins of the HT22 neuron cells were collected after OGD treatment for 4 hr and reoxygenate for 24 hr. The expression of PARP16 was detected by Western blot. After OGD/R, the expression of PARP16 increased significantly (Fig. S1A). Then, we tested the brain tissue samples of mice injured by MCAO and found that after MCAO injury, the expression of PARP16 also increased significantly (Fig. S1B). Subsequently, the expression of PARP16 were detected by immunofluorescence staning in OGD/R-treated HT22 neuron cells (Fig. S1C), and in vivo cerebral tissues were processed for immunofluorescent doubly staining using the antibody against neuron specific marker Map-2 and PARP16 (Figure S1D). The result was consistent with the protein expression, suggesting that PARP16 contributes to ER stress in cerebral I/R injury.
Knockdown PAPRP16 reduces neuronal apoptosis after OGD/R
The well-growing HT22 cells were co-incubated with PARP16 siRNA for 24 h to knockdown PARP16. After that, HT22 cells were starved in serum-free medium for 12 h and then injured by OGD/R. The control group and the OGD/R treatment group were incubated with Control siRNA (siNC) for control. The results were shown in Fig. 1A, after OGD/R, the expressions of pro-apoptotic proteins (such as p53, Bax, cleaved-Caspase 3, cleaved-Caspase 9, cleaved-PARP1) were up-regulated. After the treatment with PARP16 siRNA, the changes of protein expression were obviously reversed. Immunofluorescence analysis further showed that PARP16 siRNA pretreatment could significantly restored down-regulated Bcl-2 expression after OGD/R (Fig.1B). Subsequently, we tested the mRNA expressions of apoptosis-related genes, and the results showed that knockdown PAPRP16 could significantly inhibit the increase in the expression of apoptosis-related genes Bax and p53 induced by OGD/R (Fig. 1C). The result suggests that PARP16 contributes to ER stress-mediated apoptosis in cerebral I/R injury.
Knockdown PAPRP16 reduces neuronal inflammation response after OGD/R
Inflammation plays significant roles in the pathogenesis of ischemic stroke, and it can be regulated by intracellular multiprotein complex inflammasome (Jin et al. 2013). During ischemic brain injury, inflammasome activates Caspase-1, further leading to production of proinflammatory IL-1β and IL-18, as well as the induction of pyroptosis (Trendelenburg 2014). The expressions of Inflammation-related proteins were detected by western blot. The results showed that after OGD/R, the expressions of NLRP3 inflammatory body-associated protein (p20, cleaved-IL-1β, IL-18 and GSDMD) and inflammation-associated protein (COX-2, iNOS and VCAM-1) increased, but knockdown PARP16 could significantly reverse the expression change (Fig. 2A). Immunofluorescence analysis showed that PARP16 siRNA pretreatment could significantly down-regulate IL-1β expression in OGD/R-teated HT22 cells (Fig. 2B). Subsequently, the mRNA expressions of inflammation-related genes were tested, and the results showed that konockdown PARP16 could significantly inhibit the increase in the expression of inflammation-related genes Nos2 and Vcam1 induced by OGD/R (Fig. 2C). The result suggests that PARP16 contributes to inflammation response in cerebral I/R injury.
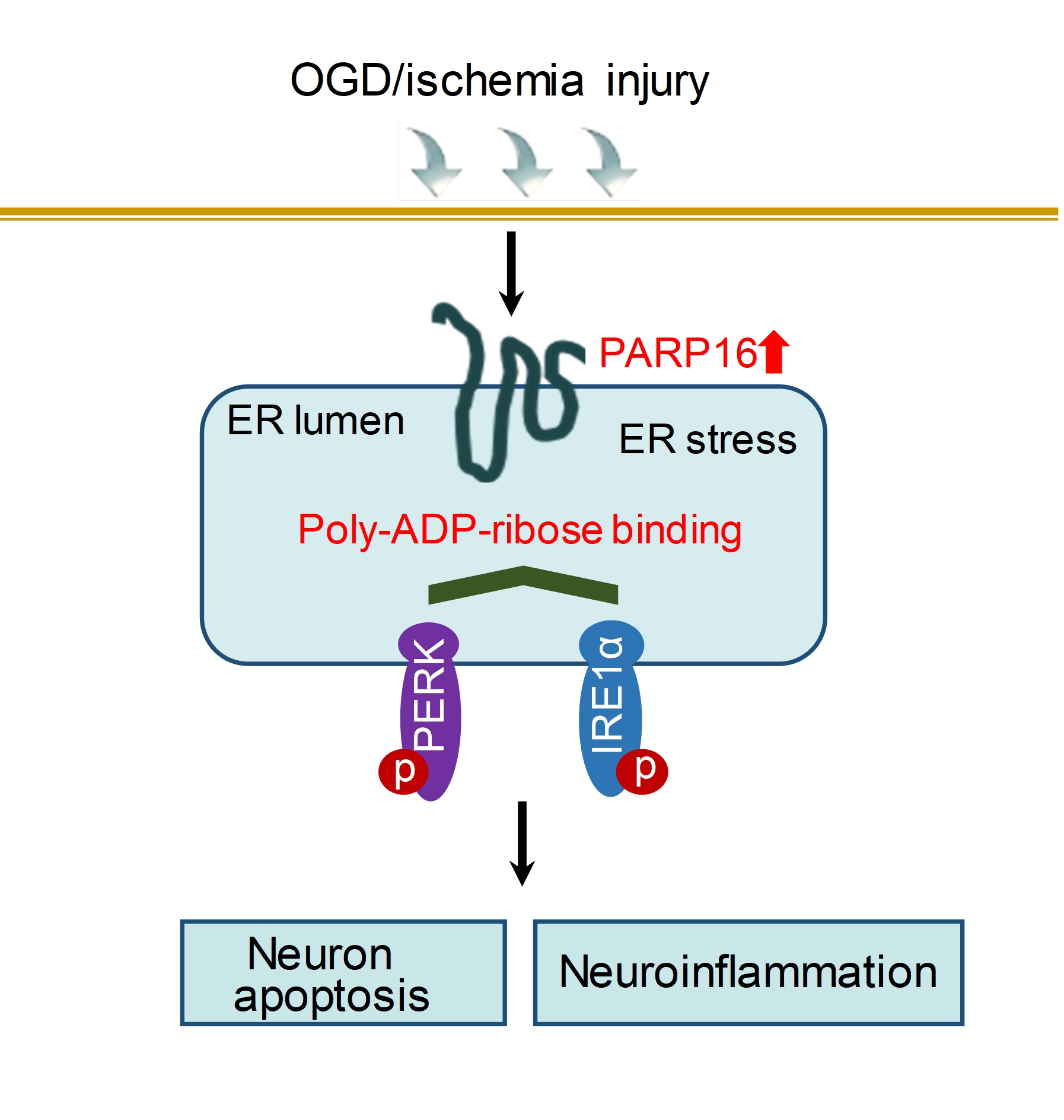
Knockdown PAPR16 reduces neuronal ER stress through inhibiting ADP-ribosylation of PERK and IRE1α after OGD/R
At first time, we confirmed that PARP16 was silenced. Subsequently, two main signaling components of endoplasmic reticulum (such as p-IRElα, p-PERK, XBP-1, Calnexin, p-elf2α) were detected by western blot. The results showed that after OGD/R, the expressions of XBP-1, Calnexin, p-elf2α, p-IRElα, and p-PERK increased, but PARP16 siRNA pretreatment could significantly reverse those changes (Fig. 3A). Immunofluorescence analysis also showed that after OGD/R, BIP expression was up-regulated, while konockdown PARP16 could significantly down-regulate BIP expression (Fig. 3B). These results suggest that PARP16 contributes to ER-stress in OGD/R-induced HT22 hippocampal neurons.
Next, we further identified how the ER stress is activated by PARP16. Because PARP16 is an effective regulator of ER stress sensor PERK and IRE1α, to clarify the interaction between the PARP16 and ER stress sensor, cell lysates were immunoprecipitated with antibody against PARP16. Co-IP results showed that PARP16 could interact with PERK and IRE1α in OGD/R-induced HT22 cells (Fig. 3C). It is generally known that ADP-ribosylation by PARP16 directly activates PERK and IRE1α. To determine whether the ADP-ribosylation of PERK and IRE1α was upregulated under OGD/R, we used Co-IP to examine the level of ADP-ribosylation of PERK and IRE1α. Our result exhibited OGD/R resulted in an increased ADP-ribosylation of PERK and IRE1α, on the contrary, knocking down PARP16 decreased ADP-ribosylation of PERK and IRE1α, suggesting that ADP-ribosylation by PARP16 was sufficient to activate PERK and IRE1α (Fig. 3D). These results suggest that PARP16 modulates the level of ribosylation of UPR activator PERK and IRE1α involved in a pathological state associated with ischemic stroke.
Knockdown PARP16 inhibits OGD/R-induced neuronal cell injury by regulating ER stress
In order to further explore whether the protective effect of knocking down PARP16 on OGD/R-injured HT22 cells is achieved by regulating ER stress, we added ER inhibitor TUDCAT or ER activator BFA to HT22 cells. The results were shown in Fig. 4, knockdown PARP16 could significantly inhibit the expressions of NLRP3 inflammatory body-associated protein (p20, cleaved-IL-1β, IL-18 and GSDMD), inflammation-associated protein (COX-2, iNOS and VCAM-1) and pro-apoptotic related proteins (Bax, cleaved-Caspase 3, cleaved-Caspase 9, cleaved-PARP1, p53) induced by OGD/R, but an ER activator BFA present, overwhelm knockdown PARP16-mediated neuronal protection in OGD/R. On other hand, when an ER inhibitor TUDCA present, permit more obvious protection caused by knockdown of PARP16 (Fig. 4A-B). Further results showed that knockdown PARP16 could significantly inhibit the activations of two main ER signaling (such as p-IRElα, p-PERK, p-elf2α, XBP-1, Calnexin, BIP) in OGD/R-treated cells, but an ER activator BFA present, reactivated PERK and IRElα signaling pathway. On other hand, when an ER inhibitor TUDCA was added, permit more obvious ER inactivation of PERK and IRF1α caused by knockdown of PARP16 (Fig. 4C). In view of the opposite result, it has been further confirmed that knockdown of PARP16 exerts neuronal protection by regulating ER stress.
PARP16 promotes ER stress-mediated primary cortical neurons damage
To ascertain whether overexpression of PARP16 induces ER stress-mediated cell injury, lentiviral expression of PARP16 was used in primary cortical neuronal culture with an efficiency greater than 90% (Fig. 5A). In addition, the levels of two main signaling components of endoplasmic reticulum (such as p-PERK, p-IRElα, p-elf2α, XBP-1, Calnexin, BIP) protein expression increased in PARP16-overexpressed cortical neurons (Fig. 5A). The results imply that the ER stress was induced by PARP16 overexpression. Overexpression of PARP16 resulted in increased expression of pro-apoptotic proteins (such as p53, Bax, cleaved-Caspase 9, cleaved-PARP1), and decreased anti-apoptotic protein Bcl-2 (Fig. 5B). Furthermore, The expressions of NLRP3 inflammatory body-associated protein (p20, cleaved-IL-1β, IL-18) and inflammation-associated protein (COX-2, iNOS and VCAM-1) increased in PARP16-overexpressed cortical neurons (Fig. 5C). These results suggest that PARP16 promotes ER stress-mediated primary cortical neurons damage.
AAV-mediated PARP16 knockdown reduces infarct formation and ER stress after MCAO in mice
To investigate the role of PARP16 in MCAO, an AAV containing an shRNA targeting Parp16 was delivered stereotaxically into the lateral ventricle. The experimental scheme was shown in Fig. 6A. Parp16 knockdown efficiency in the cortical tissue was verified by western blot analysis (Fig. 6B). Consistently, two main signaling components p-elf2α and p-PERK of endoplasmic reticulum were detected by western blot. The results showed that after MCAO, the expressions of XBP-1, Calnexin, p-elf2α, BIP, p-IRElα, and p-PERK increased, but PARP16 knockdown could significantly reverse those changes (Fig. 6C). Notably, knockdown of PARP16 decreased neurons expressing BIP protein (Fig. 6D). Furthermore, 24 h after reperfusion, neurological deficit scores and then infarction volume was measured. The results showed that pretreatment with AAV-shRNA Parp16 significantly improved the neurological deficit compared with MCAO AAV-Control group (Fig. 6E). Infarction volume was measured by TTC staining of brain tissue. As shown in Fig. 6F, the infarction volume was 39.8±1.33% in MCAO AAV-Control group. By contrast, AAV-Parp16 shRNA treatment decreased the infarction volume to 9.17±0.82%. In MCAO AAV-Control, H&E staining showed a large number of shrunken neurons with pyknotic nuclei (black arrow), which indicated dead neurons (Fig. 6G). These results suggest that AAV-mediated PARP16 knockdown reduces infarct formation and ER stress after MCAO in mice.
AAV-mediated PARP16 knockdown reduces I/R-induced apoptosis and neuroinflammation
Apoptosis and neuroinflammation is the common feature of I/R injury diseases. Western blot analysis confirmed the increased level of apoptosis in MCAO mouse brain. After MCAO, the expressions of pro-apoptotic proteins (such as Bax, cleaved-Caspase 3, cleaved-Caspase 9, cleaved-PARP1, p53) were up-regulated, but the anti-apoptotic protein Bcl-2 expression was down-regulated. After the treatment with AAV-Parp16 shRNA, the changes of protein expression were obviously reversed (Fig. 7A). Notably, knockdown of PARP16 significantly increased neurons expressing anti-apoptotic protein Bcl-2 (Fig. 7B). The expressions of inflammation-related proteins were detected by western blot. The results showed that after MCAO, the expressions of inflammation-associated protein (COX-2, iNOS, VCAM-1 and IL-6) (Fig. 7C). Immunohistochemical staining further showed that knockdown of PARP16 reduced the expression of iNOS in brain of MCAO-treated mice (Fig. 7D). NLRP3 inflammatory body-associated proteins (p20, cleaved-IL-1β, IL-18 and GSDMD) were increased in brain after MACO, but PARP16 knockdown could significantly reverse the expression change (Fig. 7E). Furthermore, breakdown of the blood-brain barrier (BBB) has been reported in stroke pathology (Khatri et al. 2012). We examined the effect of knockdown PARP16 on the expression of some proteins important to tight junction structure and function, including plasma protein ZO-1 and membrane protein Claudin-1. As shown in Fig. 7F, TJs-associated proteins decreased markedly in the damaged hemisphere, but knockdown PARP16 reversed the expression of Claudin-1 and ZO-1. These results suggest that PARP16 knockdown reduces I/R-induced apoptosis and neuroinflammation.
{kind=link}