Tissue Collection and Ethic Statement
Clinical material were obtained from patients treated at the Third Xiangya Hospital of Central South University (Changsha China) with informed consent and approval of the Medical Ethics Central South University.Tissue specimens were snap frozen and kept in liquid nitrogen until further use.
Cell lines and cell culture
All colorectal cancer cell lines were purchased from KeyGEN BioTECH (Jiangsu, China). FHC was purchased from American Type Culture Collection(ATCC) (Manassas, Virginia, USA). All cell lines had been authenticated using STR (or SNP) profiling within the last 3 years. All experiments were performed with mycoplasma-free cells.Colorectal cancer cells SW620 (RRID: CVCL_0547) and SW480 (RRID: CVCL_0546) cells were cultured in L15 (KeyGEN BioTECH, Jiangsu, China) medium supplemented with 10% fetal bovine serum (FBS, Biological Industries, Israel) and 1% antibiotics (100 U/ml penicillin and 100 mg/ml streptomycin; Life Technologies, Inc., Grand Island, NY, USA).HCT116 (RRID: CVCL_0291) and HT29 (RRID: CVCL_0320)cells were cultured in McCoy’s 5 A (KeyGEN BioTECH) medium supplemented with 10% FBS and 1% antibiotics. LoVo(RRID:CVCL_0399) cell was cultured in Dulbecco’s modified Eagle’s medium (DMEM,KeyGEN BioTECH) supplemented with 10% FBS and 1% antibiotics. FHC(RRID:CVCL_3688) cell was cultured in Roswell Park Memorial Institute 1640(RPMI 1640,KeyGEN BioTECH) medium supplemented with 10% fetal bovine serum and 1% antibiotics.All cell lines were grown in a 5% CO2 cell culture incubator at 37◦C.
Patients and tissue sampling
All clinical material were obtained from patients who underwent surgical resection for CRC at the Third XiangYa Hospital of Central South University (Changsha, China) after informed consent and approval of the Medical Ethics Central South University.
Quantitative real-time PCR assays
Total RNA from cells and tissues was extracted by TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). cDNA was generated by ReverTra Ace qPCR RT Master Mix (TOYOBO, Osaka, Japan). Quantitative real-time PCR (qRT-PCR) was carried out on a LightCycler 480 Real Time PCR instrument (Roche, Basel, Switzerland).GAPDH was used for normalization of qRT-PCR data. All primer pairs were purchased from Sangon Biotech (Shanghai, China), and sequences are available in Supplementary Table1.
Western blot assays
Whole-cells and tissues were collected and lysed with 1 × RIPA buffer (KeyGEN BioTECH) containing 1% PMSF (KeyGEN BioTECH) to harvest proteins. The protein samples were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE),and then transferred to polyvinylidene fluoride membranes (PVDF)(Millipore,CA,USA),blocked with 5% skim milk for 2h,then the PVDF membranes were incubated with primary antibodies at 4◦C overnight and secondary antibodies for 1 h, and visualized on an Odyssey CLx Infrared Imaging System (LI-COR Biosciences, NE, USA).The antibodies used for western blot (WB) were provided in Supplementary Table2.
Lentiviral Vector and Transfection
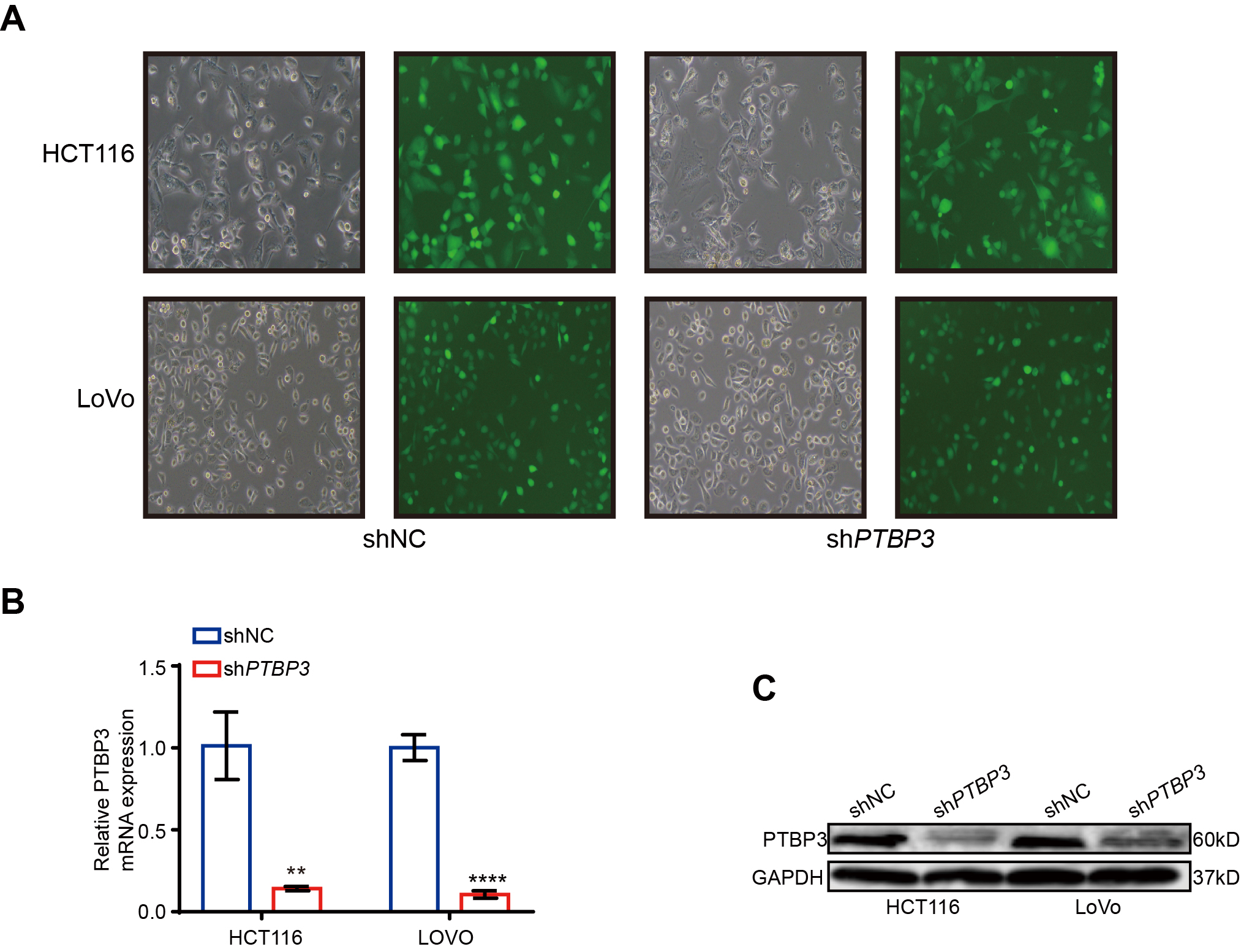
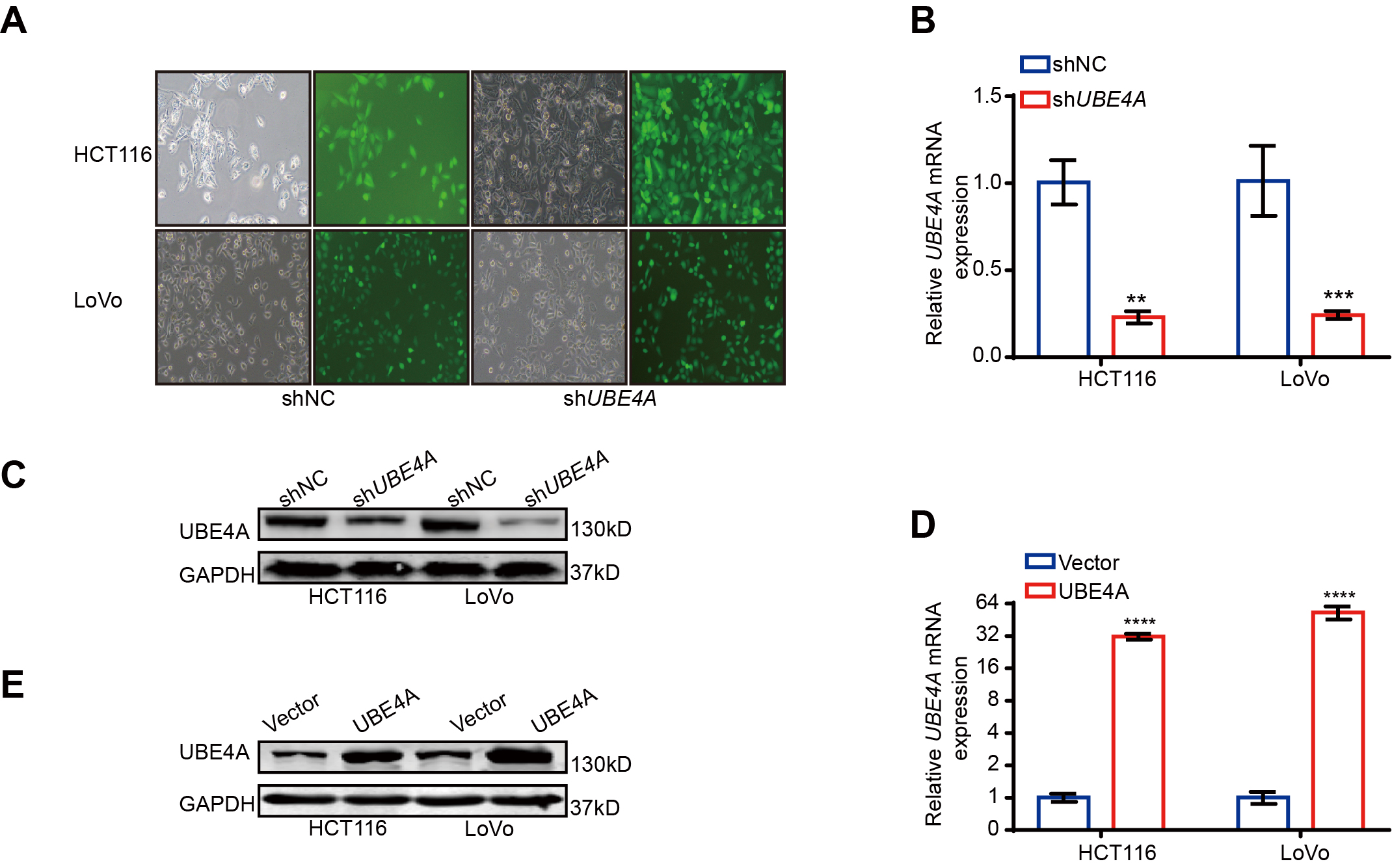
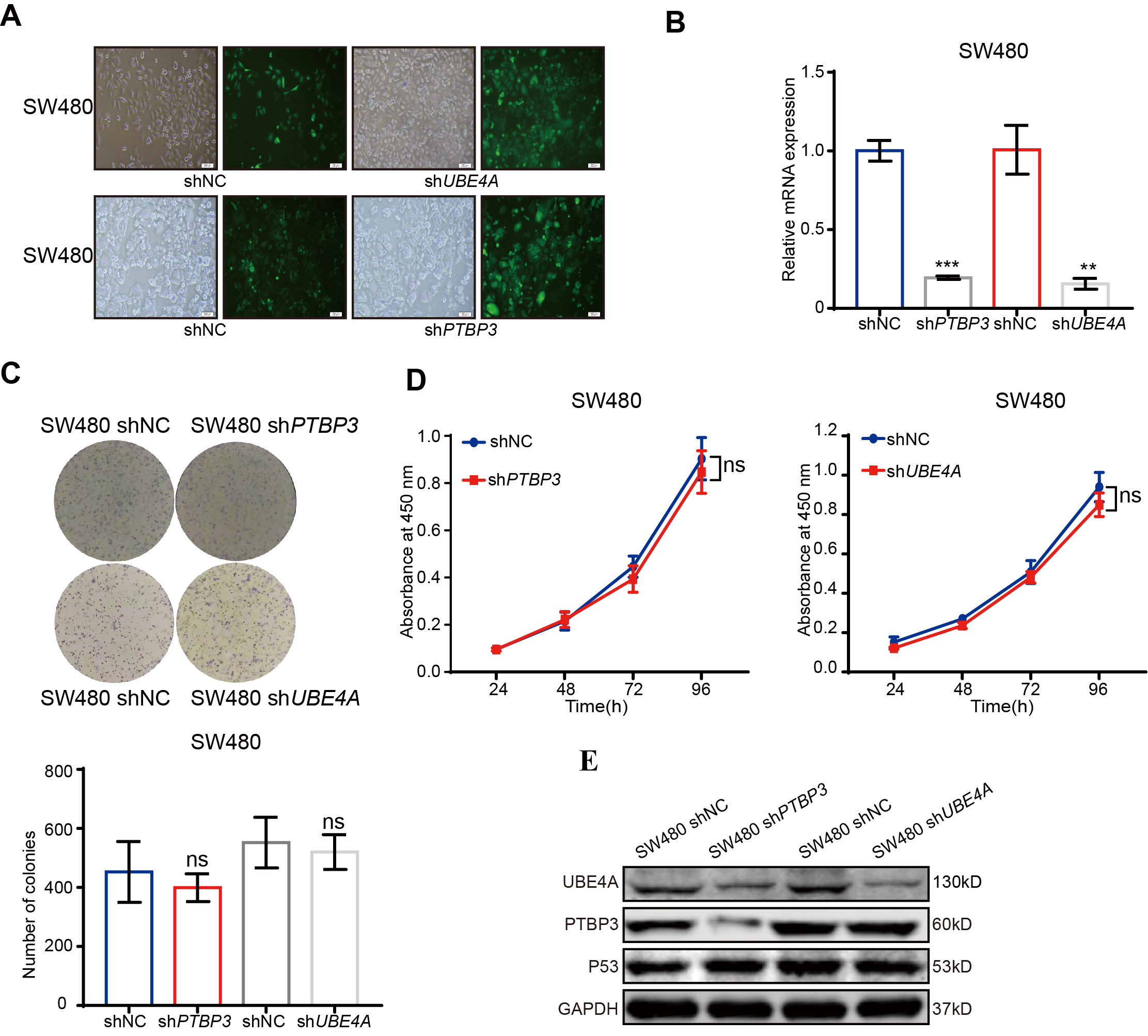
Lentiviruses, including PTBP3 and UBE4A, and their corresponding negtive control,were obtained from Shanghai GenePharma Co., Ltd. To generate stable lentivirus-transduced lines, cells were infected with virus and polybrene following the manufacturer’s recommendations, and stable cell lines were selected with 4µg/ml of puromycin treatment after 72 h of transfection. The efficiency in different cells was determined by the GFP intensity ,qRT-PCR and WB.The shRNA sequences are listed in Supplemental Table 1.
Plasmid Transfection
UBE4A plasmid were obtained from Shanghai GenePharma Co., Ltd.When tranfected the UBE4A plasmid,cells were seeded into 6-well plates and cultured for 24 hours. When the density reached 50–60%, Lipofectamine 3000 reagent (Invitrogen, USA) was used to transfect the UBE4A plasmids according to the instructions.
Cell proliferation and colony formation assays
The CCK8 assay (Dojindo, Kumamoto, Japan) was used to measure cell proliferation in 96- well plates. About 2000 HCT116 cells、2000SW480 cells and 1000 LOVO cells were seeded at per well,with six replicates for each condition. CCK8 was added at 0,24, 48, and 72 h and incubated at 37 °C for 2 h.The absorbance values (A450) were detected using an EnVision microplate reader (PerkinElmer). For colony formation assays , about 100 HCT116 cells、1000 LOVO cells and 1000 SW480 cells were seeded in each well of a 6-well plate in triplicate for each condition and incubated for 7 to 12 days. The colonies were fixed with methanol, stained with crystal violet, and counted. The average colony counts were calculated, and a paired t-test was used to test statistical signifificance. Each experiment was repeated three times.
Flow Cytometric Analysis
Cell cycle analysis was measured by flow cytometry. In general, each group prepared one hundred thousand cells labeled with propidium iodide (PI; Sigma-Aldrich, USA) was analyzed by using FACSCalibur flow cytometer (BD Biosciences). Then the proportions of G0/G1, S and G2/M cells were calculated and compared by using ModFit LT 3.1 software. The results were analyzed by using FACSCalibur flow cytometer (BD Biosciences).
Tumor Xenografts
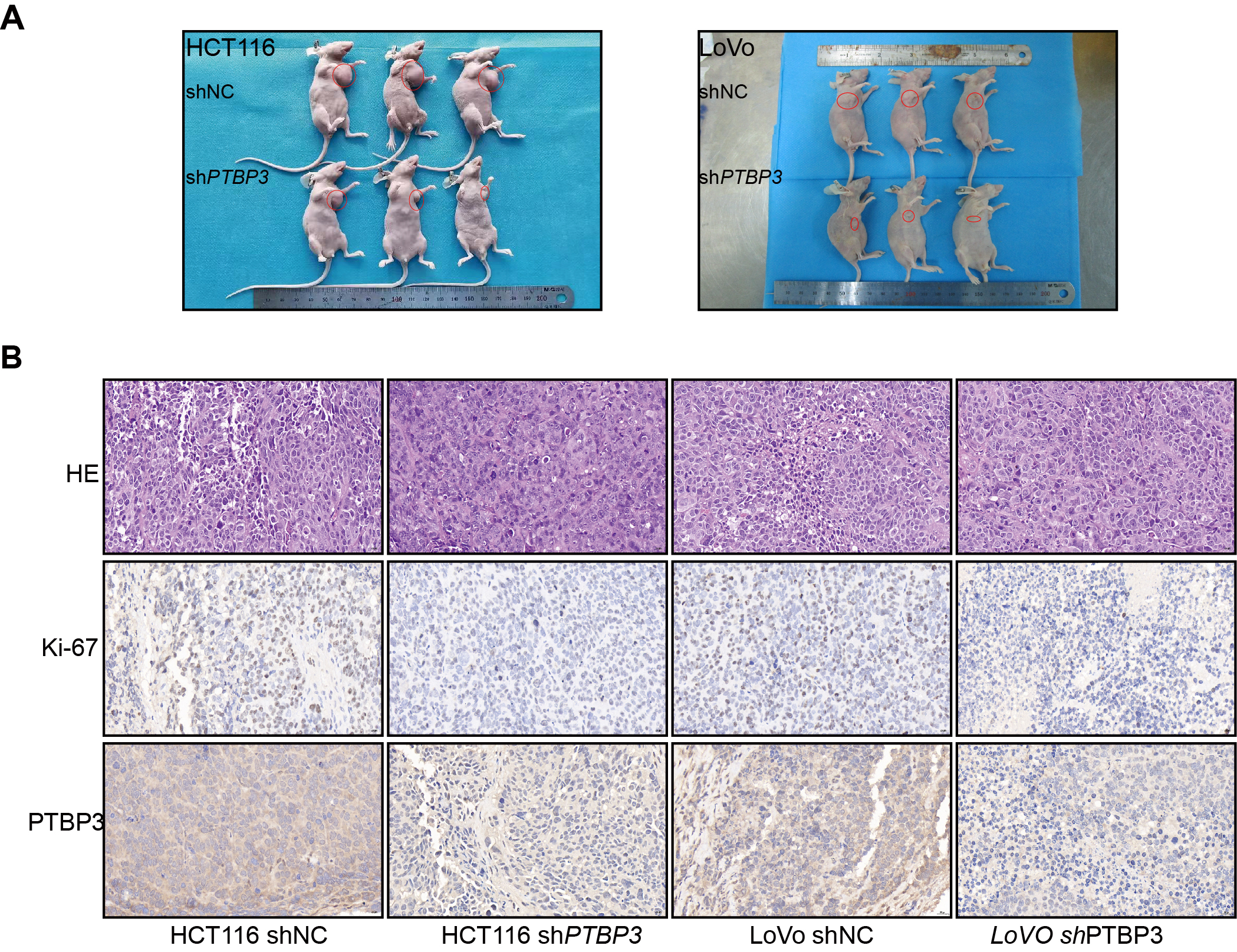
The nude mouse xenograft tumor growth model experiment was performed according to the guidelines for experimental animal management established by Kagawa University and guidelines for the welfare and use of animals in cancer research(21).In general,the Female BALB/c nude mice (4–5 weeks, 18–20 g) from the Department of Laboratory Animals of Central South University and maintained them under specifific pathogen-free conditions.Then 3 × 106 HCT116-shNC(small hairpin carrying negtive control RNAs) cells and HCT116-shPTBP3(small hairpin carrying PTBP3-specifific RNAs) cells and 2× 106 LoVo-shNC cells and LoVo-shPTBP3 cells were harvested and injected subcutaneously into the left or right flank of nude mice (n = 3 per group).Recorded the tumors with calipers and an electronic scale to estimate the tumor volume and weight every four days.After 30 days of the injection, the mice were killed by an overdose of pentobarbital (250 mg/kg, intraperitoneal injection), recorded the final results of the tumor volume and weight. The tumor volumes were calculated based on the formula: volume (mm3) = length (mm) × width (mm) × width (mm)/2. Tumors were further embedded in paraffifin for H&E and immunohistochemistry (IHC).
Immunohistochemistry
The tissue sections from the nude mouse was embedded by paraffined. Tissue sections were deparaffined and rehydrated. Blocked the tissue sections’ endogenous peroxidase activity with 0.3% hydrogen peroxide for 20 minutes. Then the tissue sections were blocked in 10% BSA for 10 minutes, and incubated with anti-human Ki-67(1:100) antibodies and anti-human PTBP3 antibody (1:100) at 4◦C for 12 h. After that tumor sections were incubated in biotinylated secondary antibodies for 20 minutes at room temperature. Thereafter, the tissue sections were reacted with streptavidin-peroxidase conjugate for10 minutes. Then added 3,3’-diaminobenzidine as the chromogen substrate. Images were captured using an inverted microscope system (Olympus, IX73, Japan).
RIP Assay
The RNA-binding protein immunoprecipitation (RIP) was performed using the EZ-Magna RIP Kit (Merck, KGaA, Darmstadt, Germany, Catalog No. 17-701) according to the manufacturer’s instructions. In general,about 2 × 107 HCT116 and LOVO cells were washed with ice-cold PBS and resuspended with RIP lysis buffer containing protease inhibitor mixture and RNase inhibitor. Then, Magnetic Beads Protein A/G was incubated with 5 µg IgG (negative control) (Merck KGaA) or PTBP3 (SantaCruz) antibody for 30 min at room temperature. Then, the complexes were added cell lysis buffer and immunoprecipitation buffer with EDTA and RNase inhibitor. Thereafter, the complexes were incubated in rotator overnight at 4◦C. The next day, the complex was washed with washing buffer containing with proteinase K and 10%SDS and then heated at 55◦C for 30 min. Finally, RNA was extracted and purified for RT-qPCR analysis. RIP assays were performed in biological triplicates and were detected by RT-qPCR ,the primers are described in Supplemental Table 1.
RNA Antisense Purification Assay
The RNA antisense purification assay was performed using the RNA Antisense Purification(RAP) Kit(Bersin BioTM ,guangzhou,China,CataLog Bes5103-3) according to the manufacturer’s instructions. In general,about 2 × 107 HCT116 cells were washed with ice-cold PBS and resuspended with methanol and 1.375 M Glycine for cross-link cells.Then added the lysis buffer containing protease inhibitor mixture and RNase inhibitor for homogenization.Next added the DNase salt stock、DNase、EDTA、EGTA、DTT for removing DNA.Then added the probes to the processed sample and hybridization at 37℃ for 30 min(the probes are desecribed in Supplemental Table1),incubate degeneration at 50 ℃ for 50 min,hybridization at 37℃ for 180 min one more time.Next added the Streptavidin Beads to the complex incubated for 30 min.After washing with wash buffer, the RAP mix bound to the beads was eluted and then re-suspended in 60μl of 1× loading buffer and boiled for 5 min, and followed with Western blot detection.
Luciferase reporter assays
HCT116sh-NC cells、HCT116sh-PTBP3 cells、Lovosh-nc cells and Lovosh-PTBP3 cells were seeded at a density of 1× 105cells per well in a 24-well plate. Cells were transfected with the pRL-TK (Promega) renilla plasmid by using Lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen). The Renilla luciferase sequence in the pRL-TK vector (Promega, WI, USA) was used as an internal control. The dual luciferase reporter assays were performed according to the protocol using the Dual-Luciferase Reporter Assay System (cat. E1910, Promega). The firefly luciferase activity was normalized to the Renilla luciferase activity. The data are expressed as the percent of luciferase activity in control cells (100%).
Immunoprecipitation Assay
The immunoprecipiton assay was perfermed according to the manufacturer’s instructions using the (Thermo,PierceTM Classic Magnetic,Rockford,USA,LOT:UC283101). In general, HCT116 cell lysates were prepared in immunoprecipitation lysis buffer (20 mM Tris-Cl, pH 8.0, 10 mM NaCl, 1mM EDTA, 0.5% NP-40) containing a protease inhibitor cocktail (Sigma). 2mg cell extracts were pre-cleared with 50μl protein A/G-agarose (Santa Cruz) at 4°C for 2h, and the supernatant was incubated with corresponding antibodies with gently shacking at 4°C overnight, followed by the addition of 50μl of protein A/G-agarose for another 2h. The beads were washed and then re-suspended in 60μl of 1× loading buffer and boiled for 5 min, and followed with Western blot detection.
Bioinformatics analysis
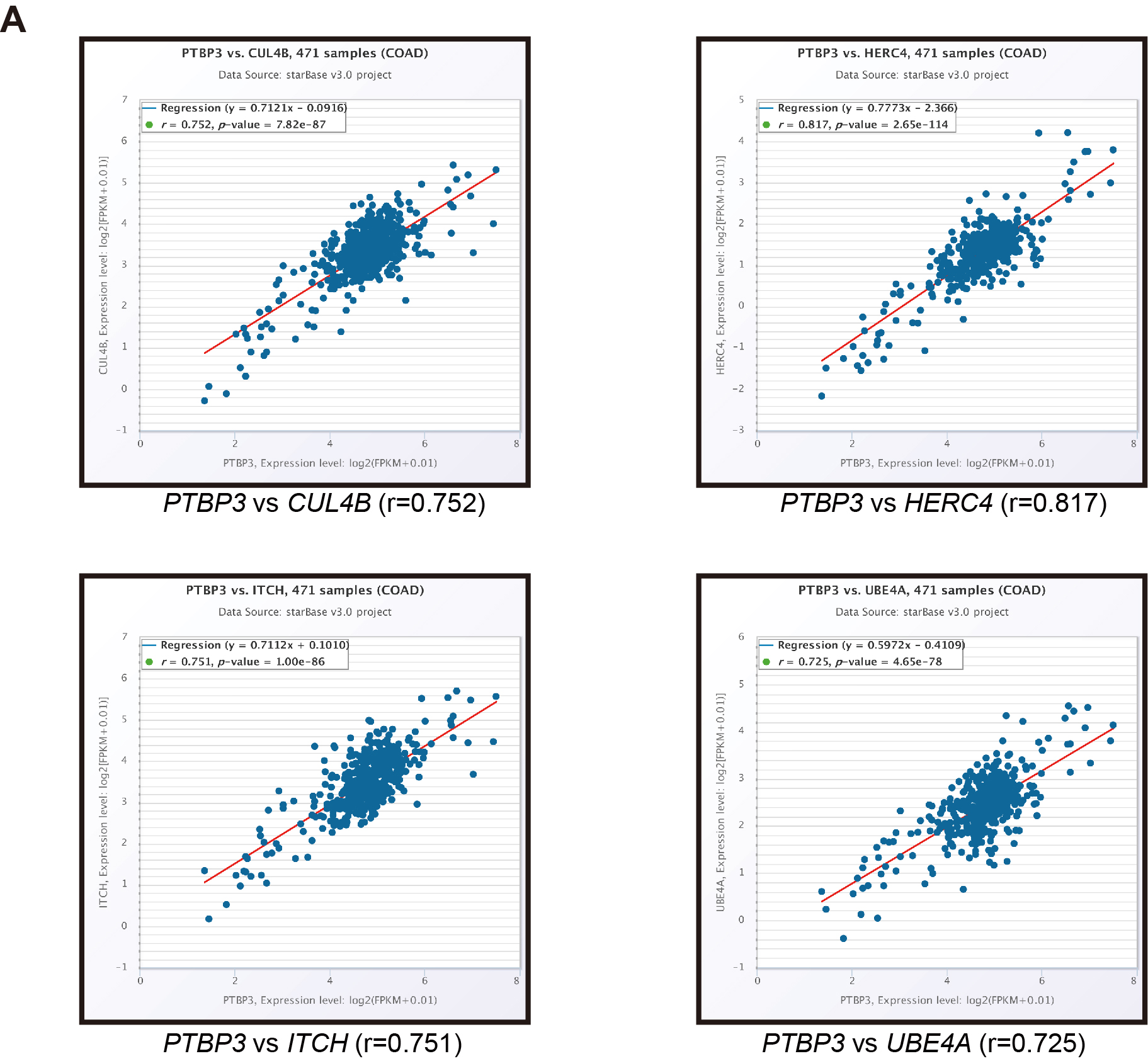
The data of PTBP3 expression analysis was downloaded from the TCGA(https://gdc.cancer.gov/)and GEO( https://www.ncbi.nlm.nih.gov/geo). The survival analysis of PTBP3 was carried out using the Gene Expression Profifiling Interaction Analysis database(http://gepia.cancer-pku.cn/)(22).The correlated gene of PTBP3 was analyzed by the TCGA data and starBase v2.0 (http://starbase.sysu.edu.cn/index.php) (23).The correlated pathway of PTBP3 analyzed by the Gene Set Enrichment Analysis(24).
Statistical analysis
Statistical computations was performed using GraphPad Prism 8.0. Data (San Diego, CA) were presented as mean ± s.d. of three independent experiments except where otherwise indicated. To compare the differences between two groups, Student’s t-test was performed.One-way analysis of variance (ANOVA) was used for comparison between the different groups. The relationship between gene expression and clinical pathological indicators is examined through chi-square test,and p < 0.05 was considered statistically signifificant.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}