Identification and validation of cancer-specific differential methylation of CpG sites
To explore NSCLC-specific DNA methylation markers, we first analyzed Infinium 450K methylation profiles obtained from The Cancer Genome Atlas (TCGA) (Fig. 1). We hypothesized that the most appropriate methylation differences (LUSC versus LUAD versus Normal) will lead to the best performance in both clustering and classification. Therefore, we started with 5 cutoff parameters of beta difference (greater than a cutoff) 0.10, 0.15, 0.20, 0.25, and 0.30 among LUAD versus LUSC versus Normal. The same cutoff parameters were utilized for LUAD and LUSC compared to normal controls. Four groups of features with each fixed parameter set were identified according to whether the difference in methylation met the cutoff: ① LUAD_LUSC_Normal specific; ② LUAD_specific; ③ LUSC_specific; and ④ Normal_specific. We assessed 4 combinations of specific probes (①; ①+②+③; ①+②+③+④; ①+④) under each fixed parameter set, resulting in 5x5x4 = 100 mixed samples. Finally, we obtained the optimal parameters for LUAD_LUSC_diff ≥ 0.20; LUAD_Normal_diff ≥ 0.10; LUSC_Normal_diff > 0.10; P < 0.05. The clustering results are shown in Fig. 2a.
Under the optimal parameters, we detected 5426 differentially methylated CpG sites (DMCs), including 5426 LUAD-LUSC cancer-specific DMCs, 1409 LUAD-normal specific DMCs and 2919 LUSC-normal specific DMCs (Supplementary Table 2). Based on differential methylation of CpG sites, we were able to distinguish LUAD, LUSC and normal tissues with diagnostic accuracies of 97.5%, 95.7% and 100%, respectively (Fig. 2b and Supplementary Table 3).
To assess the diagnostic accuracy of the methylation marker panel, we then applied the methylation panel to 1/3 of TCGA validation cohort 1 and GEO validation cohort 2 (Fig. 2c and d). The diagnostic accuracy of the panel for LUAD, LUSC and normal tissues was 98.1%, 94.8% and 100%, respectively, in 1/3 of TCGA validation cohort 1 (Fig. 2c and Supplementary Table 4) and 86.2%, 87.5% and 98.6% in GEO validation cohort 2 (Fig. 2d and Supplementary Table 5), respectively. These results demonstrate the robust nature of the methylation panel in identifying the presence of malignancy and its NSCLC subtype classification.
Cancer-specific methylation signature validation in tissues and CTCs
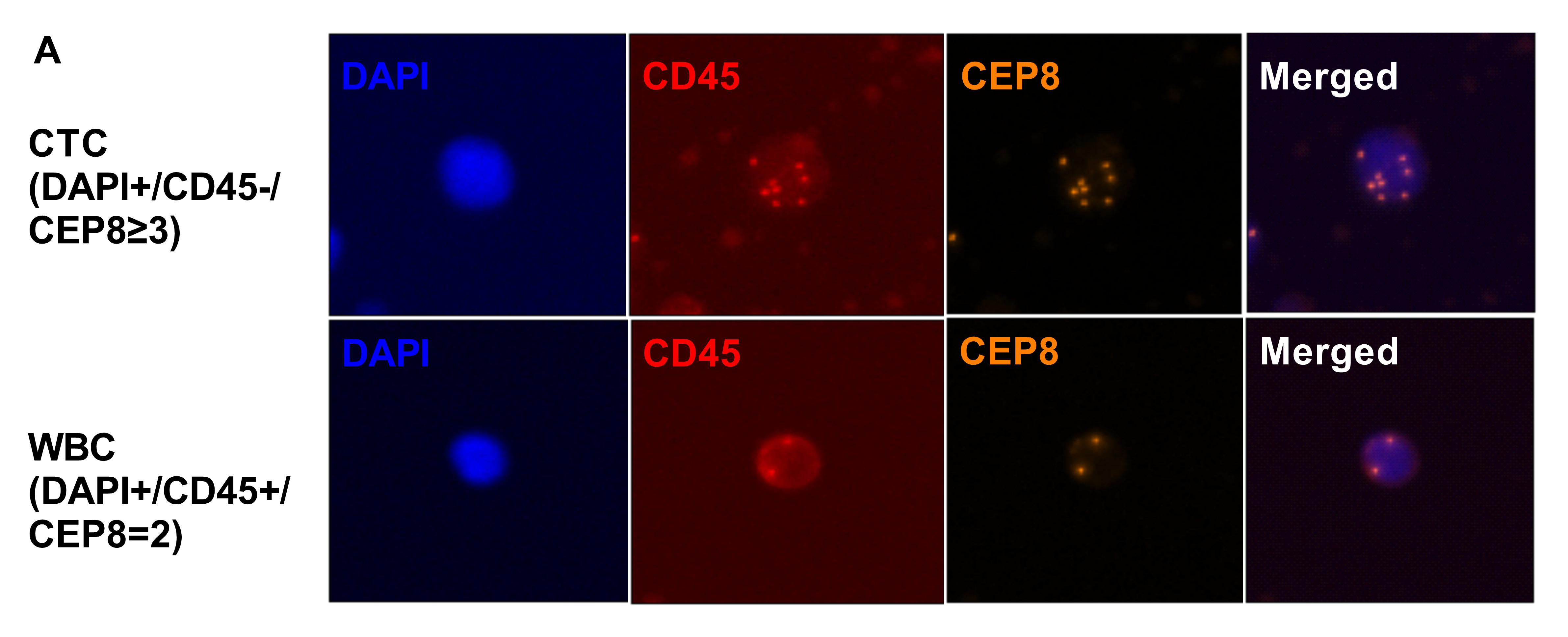
To validate the clinical value of the methylation panel, we next performed whole-genome bisulfite sequencing (WGBS) analysis of tumor tissue samples, matched normal lung tissue samples, CTCs and WBCs from 6 NSCLC patients, including 4 LUAD and 2 LUSC cases (Supplementary Table 1). To obtain CTCs, blood samples were drawn from the 6 patients with newly diagnosed lung cancer and processed with the immunostaining-FISH-based CTC technique [18–21], a CD45-based immunomagnetic system that combines leukocyte common antigen (CD45) immunostaining and fluorescence in situ hybridization using unprocessed blood samples that is specifically adapted to achieve a capture rate of > 98% for single CTC and CTC clusters [18, 22]. Upon capture, fixed CTCs were stained with antibodies against CD45 to identify contaminating leukocytes (Supplementary Fig. 1). Upon staining verification, we identified CTCs in the six patients; the CTCs from each patient were individually captured and deposited in 10 µl lysis buffer for WGBS [18, 22]. The WGBS sequencing data comprised 30G raw data (10×) for the tissue samples and bulk WBCs and 90G raw data (30×) for the CTC samples. On average, we achieved 47.3% CpG coverage for CTCs, in line with a recent single-cell WGBS study [12]. For each individual methylation profile, only CpG sites ≥ 3× coverage were used for clustering and functional analyses (Supplementary Table 6).
To refine the tumor methylation signature in the matched CTCs, we identified similar methylation CpGs (SMCs) with methylation differences < 10% and P values > 0.05 between CTCs and the matched tumor tissue for each patient and included 450K CpG sites (Supplementary Table 7). Therefore, we obtained CT450K SMCs s present in both the CTC and tumor methylation profiles with similar methylation levels that were also included in the 450K CpG sites. Then, we merged the CT450K SMCs from the 6 patients and used these CpG sites as features to cluster matched normal and lung cancer tissues, WBCs and CTCs. After refinement of the CT450K methylation signatures, all 6 pairs of CTCs and matched tumor tissues clustered together, and the Pearson correlation coefficient of CTCs and tumor tissues for each patient was greater than 0.994 (Fig. 3b). These results suggest that CTCs inherit most of their methylation signatures from the primary tumors.
However, clustering of the CT450K methylation analysis did not group the CTCs and tumors together. The high Pearson correlation coefficient of CTCs and WBCs/Normal led us to hypothesize the existence of WBCs and normal tissue backgrounds in the CTC methylation pattern. To eliminate WBC backgrounds in the CTC methylation profile, we identified CBT450K DMCs for each patient (Supplementary Table 7). Next, we merged the CBT450K DMCs of the 6 patients together and used these merged DMCs to cluster the lung cancer tissues and matched normal tissues/WBCs/CTCs. The Pearson correlation coefficient of the CTCs and WBCs for each patient decreased from a minimum of 0.831 (Fig. 3b) to a maximum of -0.696 (Fig. 3c). The results showed that almost all 6 pairs of CTCs and matched tumors clustered together, except for the clustering of 2T and 2C due to ineffective bisulfite conversion of 2B (conversion ratio 91.46%). Figure 3c reveals that the WBC background is an important component of the CTC methylation pattern.
To further eliminate the normal tissue background from the CTC methylation profile, we identified CBTN450K DMCs in each patient (Supplementary Table 7) and then merged the CBTN450K DMCs of the 6 patients together and used these merged DMCs to cluster the matched normal and lung cancer tissues, WBCs and CTCs (Fig. 3d). The poor clustering performance suggested that a normal tissue background is not an effective component of the CTC methylation pattern.
Based on the above methylation profiles, our NSCLC tissue cohort showed a diagnostic accuracy of the methylation panel for all LUAD, LUSC and normal tissues of 100% (Supplementary Table 8); after removing the matched WBC background, the diagnostic accuracy for LUAD and LUSC was also 100% based on the CTC cohort (Supplementary Table 9) (C&B diff > 0.1, P < 0.05).
Inheritance of CTCs
To explore the characteristics of CTCs, we specifically investigated the methylation profile distribution according to functional genomic features between CTCs and matched tumor tissues (Fig. 4). We observed that the number of CpG sites in each CT similar group was far larger than that in the matched CT difference group (Fig. 4a), which implies that CTCs have a large proportion of methylation signatures inherited from the primary tumor. In addition, 5-methylcytosine (5-mC) was most common in transcription factor binding sites (TFBSs) and intronic and intergenic regions, accounting for 42.8%, 47.3%, and 46.0% of all CpG sites in tumors and 47.4%, 47.6%, and 42.8% of all CpG sites in CTCs, respectively (Fig. 4a). 5-mC was also commonly found in enhancers, superenhancers, promoters, and CpG shores, accounting for 5.6%, 24.8%, 5.5%, and 6.6% of all CpG sites in tumors and 6.1%, 28.6%, 9.1%, and 8.0% of all CpG sites in CTCs, respectively (Fig. 4a).
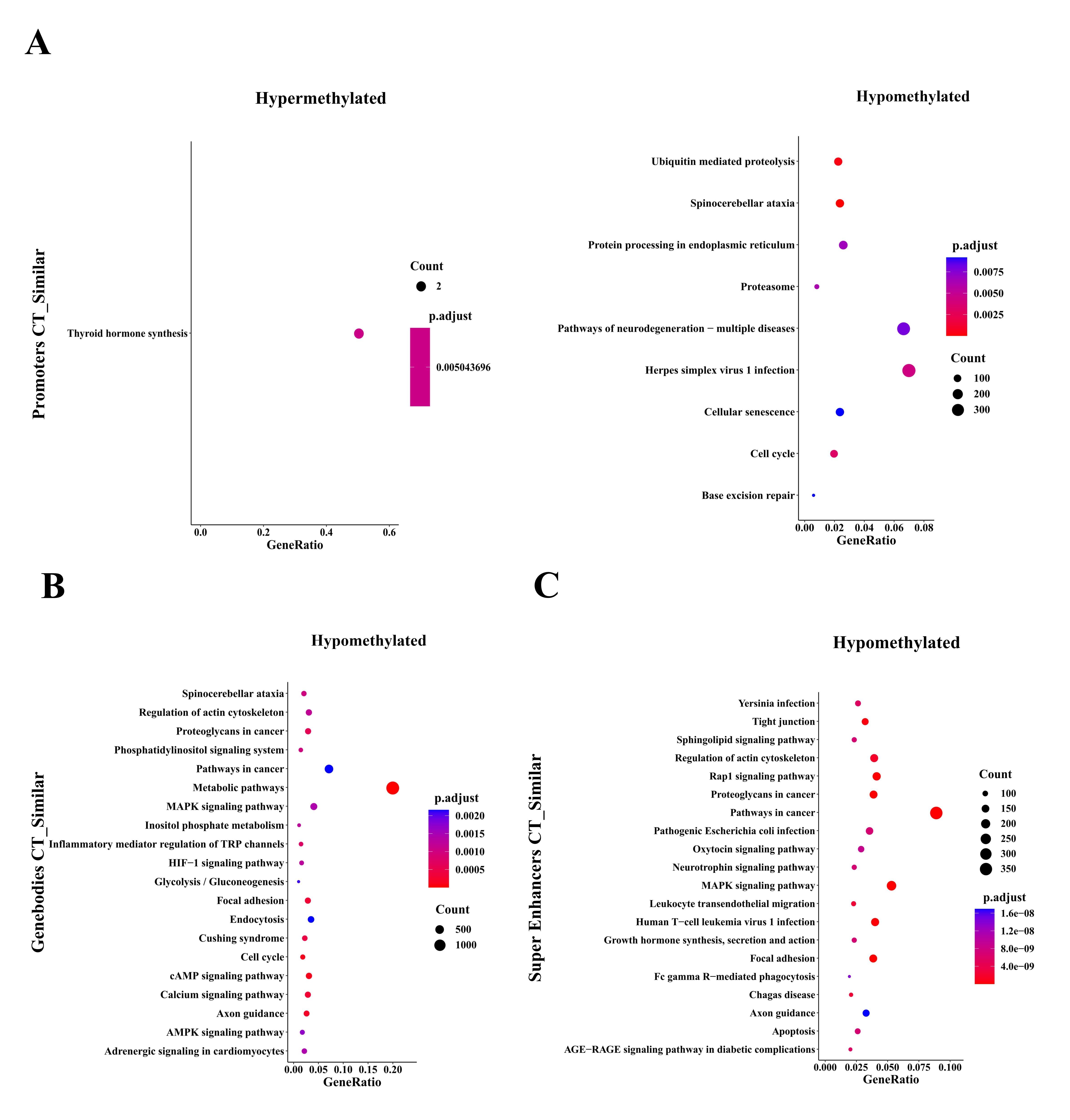
For regulatory elements, loss of DNA methylation at TFBSs can designate active transcription factor networks or networks primed for activation at later stages, e.g., during processes such as the derivation of induced pluripotent stem cells from differentiated cells [23] or cancer progression [9]. We then analyzed SMCs at TFBSs using i-Cistarget [17] and used the clusterProfile R package to analyze Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of global CTC hypomethylated TFBSs coexisting in CTCs and matched tumor tissues with methylation differences < 10% and P values > 0.05 (Fig. 5a). Our DNA methylation analysis revealed the MAPK signaling pathway and pathways regulating the pluripotency of stem cells, EGFR tyrosine kinase inhibitor resistance, and the cell cycle, among others. These pathways coexisted in both CTCs and matched tumor tissues, suggesting that CTC methylation originates from primary tumor tissues and is inherited as the cells move from the primary tumor tissues to peripheral blood (Fig. 5a). We also found that binding sites for stemness-associated transcription factors are specifically hypomethylated in SMCs in both CTCs and matched tumor tissues, including binding sites for NANOG and SOX2, which in previous reports were associated with CTC clusters compared to single CTCs [24].
To gain explore more subtle changes in DNA methylation occurring specifically within promoters, gene bodies, and superenhancer regions, we carried out hypergeometric-based gene set enrichment analysis of genomic features. Consistently, this analysis revealed hypomethylation and cell cycle progression (Supplementary Fig. 2), as previously observed for cancer specimens with stem-like and proliferative features.
Evolution of CTCs
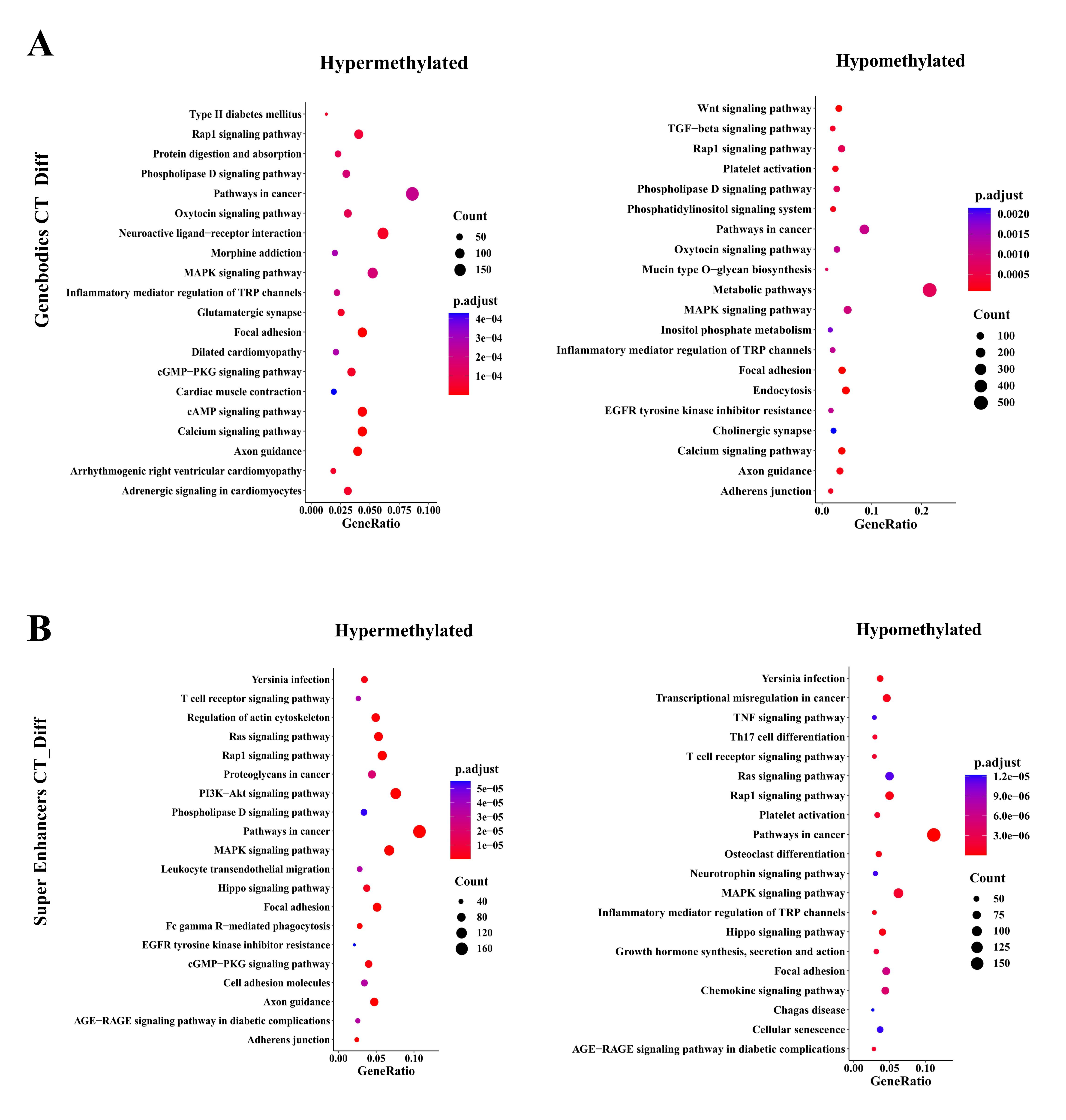
Nevertheless, CTCs showed many DMCs that differed from those of the primary tumor (Fig. 4b). To identify whether the characteristic-related transcription factor networks are also transcriptionally active in CTCs compared to matched tumors, we performed single-cell resolution methylation sequencing analysis of CTCs and matched tumors isolated from the 6 NSCLC patients (Fig. 5). We used the cluster Profile R package to analyze KEGG pathways of global hypomethylated TFBSs in CTCs rather than matched tumor tissues with absolute methylation difference > 0.1 and P value༜0.05 (Fig. 5b). KEGG analysis of TFBS DMCs in patient CTCs compared to matched tumors revealed enrichment of genes related to the T cell receptor signaling pathway, Th17 cell differentiation, TGF-beta signaling pathway, EGFR tyrosine kinase inhibitor resistance and canonical pathways (RAS/MAPK/PI3K/AKT), among others (Fig. 5b).
To gain insight into more subtle changes in DNA methylation occurring specifically within promoters, gene bodies, and superenhancer regions, KEGG analysis revealed enrichment of gene groups related to the TGF-beta signaling pathway, Th17 cell differentiation, T cell receptor signaling pathway, and EGFR tyrosine kinase inhibitor resistance, among others (Supplementary Fig. 3).
These results show that in the processes of dissemination from the primary tumor tissue to the peripheral blood, CTCs gradually develop their own unique methylation signatures with some unique characteristics different from those of the primary tumor.
{kind=link}
{kind=link}
{kind=link}