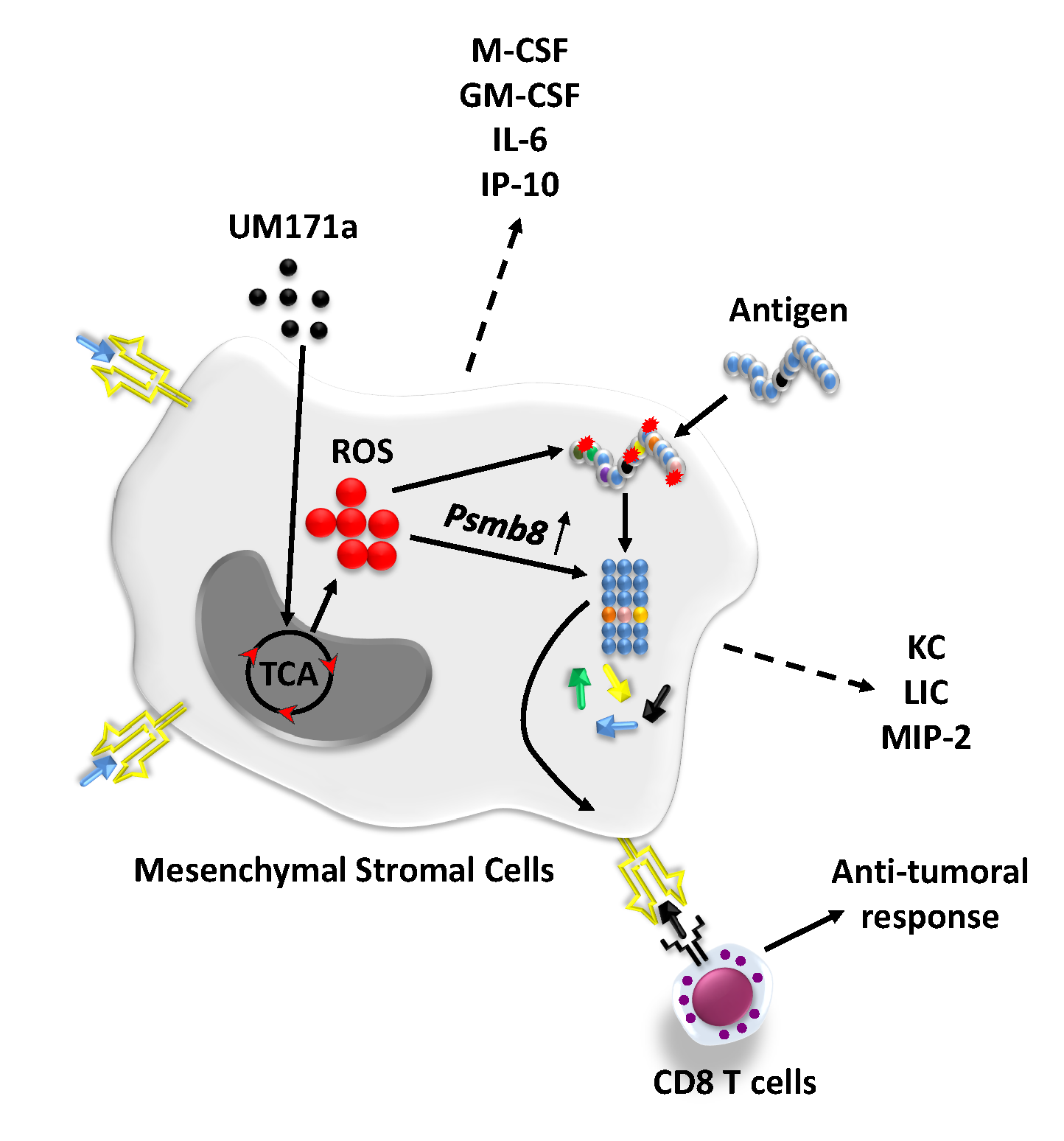
UM171a is well tolerated by primary MSCs and triggers MHCI up-regulation.
The parent UM171a compound was initially discovered by a high-throughput screening assay designed for the identification of compounds capable of triggering leukemic stem cell proliferation.19 A series of chemical modifications were then conducted to create the final UM171a product, which effectively promotes ex vivo expansion of long term (LT)-hematopoietic stem cells (HSCs).20 When further studied to decipher its potential mode of action on human CD34+ HSCs, UM171a was found to trigger a marked increase in the expression of several immune-related genes including human leukocyte antigens (HLA)-A and B - ortholog of the murine MHCI (aka H2-K/H2-D)-, beta 2-microglobulin (β2M) as well as the co-stimulatory molecule CD86.21 Since these specific genes are central to antigen presentation, we posited that treatment of primary murine MSCs with UM171a would trigger or enhance the expression of these genes resulting in the acquisition of antigen presentation properties. Prior to testing this hypothesis, we first identified the working concentration of UM171a by conducting MTD experiments on murine MSCs over three consecutive days. We elected to work with a UM171a concentration of 1000 nM as higher doses impair cell proliferation (Fig. 1A). Further characterizations revealed that UM171a treatment did not alter the innate MSC phenotype as the cells remained CD45 negative while expressing CD44, CD73, CD90.1 and CD105 (Fig. 1B). Although no increase in cell surface expression of the co-stimulatory molecules CD86 nor its homolog CD80 was detected on murine MSCs (Fig. 1B), a sharp increase in H2-Kb expression was observed (Fig. 1B). To see if this H2-Kb increase requires a 72 h treatment and/or a dose as high as 1000 nM, we evaluated the effects of multiple UM171a doses (35, 250 or 1000 nM) in a timely manner. Indeed, H2-Kb levels were only enhanced following a three-day treatment with 1000 nM of UM171a (Fig. 1C), and remained steady up to a dose of 8000 nM (Fig. 1D). Interestingly, assessment of EPCR expression, a marker of UM171a-induced activation, followed an expression profile kinetic akin to H2-Kb (Fig. 1E) indicating a direct correlation between H2-Kb increase and enhanced EPCR expression. To ensure that these observations can be replicated using human cells, human UC-derived MSCs were treated with 1000 nM UM171a and showed a similar increase pattern in HLA-A/B/C expression (Fig. 1F). Altogether, these results indicate that UM171a is well tolerated by MSCs and can trigger a potent increase in MHCI/HLA cell surface expression.
Treatment of murine MSCs with UM171a instills antigen cross-presentation abilities with no protagonist effect on antigen uptake and processing.
The observed increase in MHCI/HLA levels on the surface of UM171a-treated MSCs suggests that these cells may exhibit enhanced antigen presentation or the capacity to cross-present captured soluble antigens to responding CD8 T cells. We thus tested whether the identified dosing and treatment regimen affects antigen cross-/presentation by MSCs following soluble OVA protein or SIINFEKL peptide pulsing respectively (Fig. 2A). Besides exhibiting enhanced antigen presentation (as shown by the SIINFEKL response), UM171a-treated MSCs were also able to cross-present the immunogenic OVA-derived SIINFEKL peptide (Fig. 2B) with a comparable T-cell response following longer (7 instead of 3 days) treatment regimen (Fig. 2C-D). When tested on MEFs - another non-hematopoietic cell - UM171a treatment failed to trigger antigen cross-presentation despite improved antigen presentation (Fig. 1E) and increased EPCR expression (Fig. 1F). Since the observed antigen cross-presentation effect mediated by UM171a can be potentially enhanced by increased extracellular antigen capturing and/or intracellular processing, MSCs were first treated with UM171a for three days then pulsed with either fluorescent OVA-AF647 (to assess antigen capturing) or OVA-DQ (to evaluate OVA processing). Compared to DMSO-treated MSCs, no increase in antigen uptake (Fig. 2G) nor antigen processing (Fig. 2H) was observed. The sum of these observations stipulates that UM171a can trigger de novo antigen cross-presentation by MSCs in a mechanism(s) independent of enhanced antigen uptake or processing.
Reactive oxygen species (ROS) production drives antigen cross-presentation in UM171a-treated MSCs
Since antigen up-take and processing could not explain the induced cross-presentation ability of UM171a-treated MSCs, we next wondered whether such treatment affects the endoplasmic-reticulum (ER)-associated protein degradation (ERAD) machinery. ERAD is a cellular pathway responsible for targeting misfolded proteins for ubiquitination and subsequent degradation by the proteasomal complex.22 Analysis of publicly-available transcriptomic data conducted on human HSCs revealed UM171a-mediated changes in the expression of several ERAD-related genes such as Erap1/2, β2m, Tap1/2 as well as H2-t and H2-q molecules (Fig. 3A). Although expression of murine homolog of these genes remained steady in UM171a-treated murine MSCs, a noticeable increase in the expression of other tested genes, Psmb8 and Calr, was observed (Fig. 3B). This is a salient observation for three main reasons. First, Psmb8 - the β5i-subunit of the immunoproteasome - possesses a strong chymotryptic- and tryptic-like processing activity capable of generating 8–9 amino-acid peptide fragments that can efficiently fit within MHCI grooves.23 24 Second, Calr plays an important role in capturing misfolded proteins preventing their migration from ER to the golgi apparatus.25 Third, the expression of these two genes can be induced in response to misfolded proteins that accumulate intracellularly due to aggregations or damages inflicted by elevated ROS production.26–29 This is in line with the previous observation that treatment of human CD34+HSCs with UM171a induces detoxification pathways as a defense mechanism to counteract the toxic effects mediated by elevated ROS levels.21 When we investigated UM171a-triggered ROS (more specifically superoxide anion) production in both murine MSCs and MEFs following a 72 h treatment condition, a signal was only detected in MSCs (Fig. 3C). Production of superoxide anion production was however completely abolished in MSCs following MitoTEMPO (an inhibitor of mitochondrial-derived ROS), vitamin E derivative α-tocopherol (inhibitor of lipid peroxidation), or NAC (a general antioxidant and cysteine donor) co-treatments (Fig. 3D). These observations prompted us to further explore whether ROS production predisposes MSCs to acquire antigen cross-presentation abilities. We thus, co-treated UM171a-pulsed MSCs with the same anti-oxidants listed above prior to conducting an antigen presentation assay. As shown in Fig. 3E, addition of MitoTEMPO or α-tocopherol completely blunted antigen cross-presentation by UM171a-treated MSCs whereas significant inhibition was observed with the use of NAC. Antigen presentation (e.g. SIINFEKL pulsing), on the other hand, remained unchanged between anti-oxidants and control treatments. To further re-enforce this hypothesis, we next compared the transcript levels of Psmb8 in UM171a-treated MSCs co-treated with anti-oxidants. As expected, Psmb8 expression was impaired in response to α-tocopherol, MitoTEMPO or NAC (Fig. 3F), clearly indicating a central role played by ROS in mediating antigen cross-presentation via de novo expression of Psmb8.
ROS are generally produced by mitochondria during the process of oxidative phosphorylation.30 More specifically, electron transfer between complexes of the electron transport chain (ETC) lead to partial reduction of oxygen to form superoxide anion.31 Since UM171a triggers both ROS and their cognate detoxification mechanisms, it is logical to stipulate that it may act either directly or indirectly onto ETC complex(es). We thus tested the effect of various ETC inhibitors (ETCi - Fig. 4A) on MSC-mediated antigen cross-presentation as a co-treatment strategy with UM171a (e.g. since day 1) or during the antigen pulsing step (after the three-day treatment period with UM171a - Fig. 4B). Surprisingly, antigen cross-presentation by UM171a-treated MSCs was unaffected by ETCi during the co-treatment regimen (Fig. 4C - upper panel) whereas a significant decrease in B3Z activation was only observed when Antimycin-A (inhibitor of complex III) was co-treated with OVA (Fig. 4C - lower panel). Interestingly, UM171a-pulsed MSCs co-treated with Antimycin-A showed very low or absent superoxide anion production (Fig. 4D) with the absence of major effects on OVA uptake or processing (Fig. 4E). These results clearly indicate that mitochondrial-derived ROS production is the main factor driving antigen cross-presentation by UM171a-treated MSCs.
UM171a treatment does not induce PD-L1 expression on MSCs.
We know so far that MSC treatment with both UM171a or IFN-gamma leads to enhanced MHCI expression (Fig. 5A) and promotes antigen cross-presentation (Fig. 2B).6 We thus decided to compare the functional potency of both treatments in an antigen presentation assay. Since the OVA pulsing protocols for UM171a- and IFN-gamma-treatment are different (8 versus 18 h respectively), we tested both conditions and noted a significantly higher T-cell response with the IFN-gamma treatment (Fig. 5B) most likely owing to the elevated H2-Kb levels following IFN-gamma treatment (Fig. 5A). Interestingly however, UM171a did not induce de novo expression of PD-L1 on the surface of MSCs compared to IFN-gamma treatment (Fig. 5C). Since the B3Z cell line may not be highly responsive/sensitive to PD-1/PD-L1 interaction due to its low/absent PD-1 expression profile (small panel in Fig. 5C), we repeated the antigen presentation assay using primary OT-I-derived CD8 T cells and assessed their responsiveness by quantifying both IFN-gamma and IL-2 production. Although the T-cell response to SIINFEKL presentation by IFN-gamma-treated MSCs was substantially higher compared to the UM171a-treated group (Fig. 5D-E), the antigen cross-presentation ability of UM171a-treated MSCs was superior to IFN-gamma treatment (Fig. 5D-E), but became comparable to the IFN-gamma group in the presence of PD-L1 neutralizing antibodies (Fig. 5D-E). These results clearly highlight a therapeutic advantage for the use of UM171a as it precludes the negative role played by PD-L1 expression normally induced in response to IFN-gamma stimulation.
Therapeutic vaccination using UM171a-treated MSCs delays tumor growth.
Given the potent in vitro cross-presentation ability of UM171a-treated MSCs, we finally assessed the ability of these cells to trigger anti-tumoral immune response in immunocompetent animals with pre-established EG.7 T-cell lymphomas (Fig. 6A). The SC delivery of OVA-pulsed MSCs treated with UM171a significantly delayed tumor growth compared to OVA-pulsed MSCs or untreated control mice (Fig. 6B) with a 40% survival rate reached up to 40 days post-tumor transplantation (Fig. 6C). Although this therapeutic effect can be explained by the immunogenic potential of the vaccine (e.g. OVA-derived peptides), MSCs can further modulate immunity via their capacity to secrete various immune soluble mediators.32 33 We thus evaluated whether UM171a affects the secretome of MSCs, hence amplifying their anti-tumoral properties. Indeed, a three-day treatment with UM171a led to significant increases in various pro-inflammatory cytokines (M-CSF, GM-CSF, IL-6, and IP-10) and chemokines (KC, LIX, MIP-2 - Fig. 6D), which are all known for their ability to recruit and modulate the activity of host-derived innate and adaptive immune cells. Altogether, these findings indicate that UM171a-treated MSCs can be effectively exploited in the design of cellular vaccines capable of triggering potent anti-tumoral responses.
{kind=link}