Synthesis of Cu 6 (C 4 H 3 N 2 S) 6 . 2-Mercaptopyrimidine (≥ 99%) were purchased from Alfa Aesar. DMF and Copper nitrate trihydrate(Ⅱ) (Cu(NO)3, ≥ 99%) were bought from Sinopharm Chemical Reagent Co., Ltd., China. All reagents and solvents used were of commercially available reagent grade and used without further purification. 2-Mercaptopyrimidine (112mg, 1mmol) and Cu(NO3)2·3H2O (240mg, 1mmol) were dissolved in 20ml DMF solution respectively, stir thoroughly to dissolve completely. Subsequently, add the DMF solution of 2-Mercaptopyrimidine dropwise to the solvent of Cu(NO3)2·3H2O. Cu6L6 powder crystals (C24H18Cu6N12S6) were obtained by filtration after vigorous stirring at room temperature for two hours. (A single crystal suitable for SCXRD measurements can be obtained by reducing the dosage in the same proportion).
Characterization. Powder X-ray diffraction (PXRD) patterns of the samples were acquired on on a Riguku D/Max-2500PC X-ray diffractometer with Cu radiation (λ = 1.54178 Å). Fourier transform infrared (FT-IR) spectroscopy was conducted using a Bruker ALPHA II FT-IR spectrometer. X-ray photoelectron spectroscopy (XPS) analysis was obtained on an ESCALAB 250 instrument operated at 150 W and 200 eV with mono chromated Al Kα radiation. Thermogravimetric analyses (TGA) were performed on an SDT 2960 thermal analyzer from room temperature (RT) to 800°C at a heating rate of 10°C/min under a nitrogen atmosphere. Dynamic light scattering (DLS) data were obtained by Horiba nano Partica SZ-100V2. UV-vis absorption spectra were recorded using a Hitachi UH4150 UV-visible spectrophotometer in the range of 200–700 nm. Emission and excitation spectra at RT were recorded with an Edinburgh FLS 1000 fluorescence spectrometer, and luminescence microscopy images were recorded on an Olympus BX53 microscope. Electron paramagnetic resonance (EPR) spectra were recorded by a Bruker A 300 EPR spectrometer.
Single-Crystal X-ray Diffraction (SCXRD) analysis. SCXRD was performed on a Rigaku XtaLAB Pro diffractometer Cu-Kα radiation (λ = 1.54184 Å) at 200 K. Data collection and reduction were conducted with CrysAlisPro software. The structures were solved with intrinsic phasing methods (SHELXT-2015) and refined by full-matrix least squares on F2 using OLEX2, which utilizes the SHELXL-2015 module. The imposed restraints in least-squares refinement of each structure were commented in the corresponding CIF files. All non-hydrogen atoms were refined anisotropically, and the hydrogen atoms were included on idealized positions. The crystal structures are visualized by DIAMOND 3.2. The detailed information of the crystal data, data collection and refinement results are summarized in Table S1.
Cell Culture. Cell lines including human melanoma cells (A375), human esophageal cancer cells (Kyse30), human breast cancer cells (MCF-7) and Rat cardiomyocytes cells (H9C2) were purchased from the cell bank of the Chinese Academy of Sciences (Shanghai, China). A375 and H9C2 cells were cultured in DMEM medium containing 10% FBS and 1% penicillin-streptomycin. MCF-7 and Kyse30 cells incubated in 1640 medium containing 10% FBS and 1% penicillin-streptomycin.
Cytotoxicity Analysis. A375, MCF-7 and Kyse30 cells were seeded in 96-well plates (8×103 cells per well), respectively, and cultured for overnight. Then these cells were incubated with different concentrations of Cu6NC (0, 2, 4, 8, 12, 15, 20 and 25 µM), the incubation time was 24 hours and 48 hours, respectively. Finally, add 10 µL of CCK-8 solution to each well and incubate for another 1 h. The absorbance of cells in each well at 450 nm was measured by a microplate reader. The cytotoxicity test of Cu6NC on H9C2 cells was also carried out according to the above description.
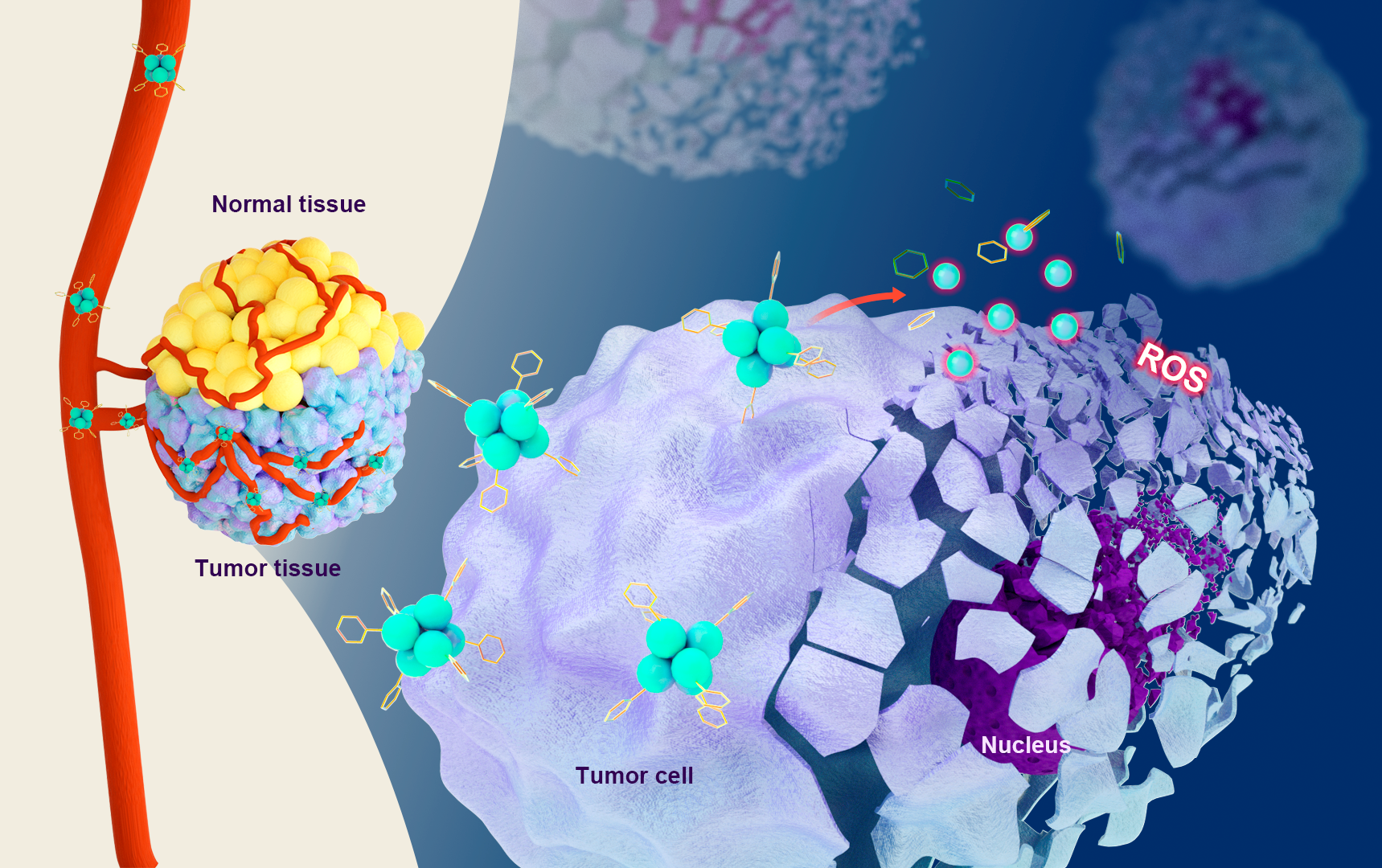
Cellular Uptake of Cu6NC
A375 and H9C2 cells were seeded in culture dishes and incubated for 24 h. Subsequently, cells were incubated with 8µM Cu6NC and harvested after 3 h and 6 h. The percentage of positive cells was tested by the flow cytometry (ACEA NovoCyte3130, USA) with an excitation wavelength of 405 nm and an emission wavelength of 780 nm. Data were processed by FlowJo software.
Apoptosis and Cell Cycle Analysis. A375 (2.5×105 cells per well) cells were seeded in 6-well plates with 0, 2, 8 and16 µM Cu6NC for 24 hours. The cells were collected using trypsin-EDTA and stained with annexin V-FITC/PI (BD Biosciences, USA) for 15 min, f and then apoptotic cells were identified by flow cytometry. The cell cycle was measured by PI staining using a cell cycle analysis kit (KeyGen Biotech, China). After staining following the manufacturer’s instructions, the cells were analyzed by flow cytometry (FACS Aria III, USA). Data were processed by ModFit LT software.
Cell Dead/Live Assay. A375 cells were treated with Cu6NC (0, 2, 8 and 16 µM) for 24 h. Before being observed with a fluorescence microscope, the cells were washed twice with PBS and incubated with 2 µM calcein AM and 4.5 µM PI for 20 minutes. For flow cytometry detection, after being digested and harvested, the cells are washed twice by PBS and then tested by a FACS Aria III flow cytometer. Data were processed and analyzed by FlowJo software.
Transwell migration assay. Migration assays of A375 in vitro were performed using a 24-well plate with a transwell polycarbonate permeable support (pore size, 8 µm; Corning Incorporated, Corning, USA). A375 cells were incubated and treated as described in the Apoptosis Assay. The harvested cells were resuspended in FBS-free medium and seeded in the upper chamber with 1×105 cells per well. The lower chamber was filled with 20% FBS medium (600 µL/well). The cells were then cultured for 48 hours. Finally, the cells in the lower layer of the upper chambers were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. Numbers of migrated cells were photographed with a fluorescence microscopy and processed by ImageJ software.
Colony Formation Ability Identification. A375 cells were incubated and treated as described in the Apoptosis Assay. The cells were digested and seeded in 6-well plates (1000 cells per well). Then the cells are cultured in complete medium, and the medium was changed every 4 days. At 12 days, the cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. The cell colonies were observed and photographed with a microscope, and the colonies containing more than 50 cells were counted using ImageJ software.
Intracellular Reactive Oxygen Specie (ROS) Generation. A375 cells (5×104 cells/well) were incubated in a 24-well plate with different concentrations of Cu6NC (0, 2, 8 and16 µM) for 12 h. Then, the level of cellular ROS was measured by 2′,7-dichlorodihydrofluorescein diacetate (DCFH-DA) probe (Beyotime Biotechnology, Jiangsu, China). Firstly, the cells were washed twice with PBS, incubated with DCFH-DA (10 µM) for 25 minutes, and then washed with PBS three times. Finally, the ROS signal of the cells was observed by a fluorescence microscopy.
Assessment of Glutathione (GSH). Levels of GSH in A375 cells after treatment with Cu6NC were evaluated with GSH and GSSG assay kits (Beyotime Biotechnology, Jiangsu, China). In short, A375 cells were seeded in 6-well dishes (2×105 cells per well) and treated with different concentrations of Cu6NC (0, 2, 8 and 16 µM) for 6 h. Then, the cells were collected with centrifugation, and the supernatant was discarded. Samples were added protein remover M (10 mg/30µL), subjected to 3 cycles of freezing − thawing, and then centrifugated at 1000 g for 10 min at 4°C. The supernatant was reserved for GSH measurement with GSH and GSSG assay kits according to the manufacturer’s protocol.
In vivo Chemodynamic Therapy. All animal procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals and were approved by the Welfare and Ethics Review Committee of Zhengzhou University Laboratory Animal Center (Approval number: ZZU-LAC20200911[14]). In order to generate a xenograft mouse model, on day 0, we planted A375 cells (5×106 tumor cells in 200 µL of PBS) in 6-week-old female BALB/c nude mice (Beijing Vital River Laboratory Animal Technology Co. Ltd, China) subcutaneously in the right flank. When the tumor volume reached about 100mm3 on the 4th day, we randomly divided the mice into three groups: normal control group (NC), Cu6 NPs group (10 mg/kg), and Cu6NC group (20 mg/kg), at the same time by intraperitoneal injection corresponding dose of Cu6 NPs to treat mice. Mice were exposed to these treatments every other day for a total of nine administrations. The length (L) and width (W) of the tumor were measured with a vernier caliper every two days, and the tumor volume (V) was calculated using the formula: V = L×W2/2. On the 20th day, the tumor tissues, peripheral blood, and major organs (heart, liver, spleen, lungs and kidneys) of mice were harvested.
All harvested tissues were fixed with 4% formalin at least for 48 hours. The fixed tissue is used for experiments such as hematoxylin and eosin (H&E) and immunohistochemistry staining. Apoptosis of tumor tissue sections was detected by staining with terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). The proliferation of tumor tissue was detected by immunohistochemical Ki67 staining. H&E were used to stain sections of major organs in each group. All these stained sections could be observed with a fluorescence microscopy. After the peripheral blood was collected, a serum sample was obtained by centrifugation. Then, an automatic biochemical analyzer (Chemray 240 or 840, Rayto, China) was used to detect the indexes of liver function, kidney function and myocardial enzymes.
Statistical Analysis. All results are presented as the mean ± standard deviation (SD). Two treatment groups were compared by Student’s t test. Multiple group comparisons were performed by two-way analysis of variance with Tukey’s post hoc test. All statistical analyses were carried out using GraphPad Prism 5. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 were deemed as significant differences. “NS” indicates no significant differences.
{kind=link}