Data available
No animal experiments were necessary for this study, therefore no ethics committee approval was required. Sequence data were obtained from the VarGoats project using Illumina HiSeq or Illumina NovaSeq technologies, the first step towards a 1,000 goat genome project (http://www.goatgenome.org/vargoats.html). The current data bank comprises 808 individuals from Capra hircus of various breeds and geographical origins, as well as 21 wild goat individuals. Forty-four French Alpine and thirty-seven French Saanen individuals were sequenced at Genoscope (Evry, France) with an average coverage of 12X. The individuals sequenced were selected to best represent the genetic structure of the current French population: AI bucks from the largest families, maximized haplotype coverage from the population by picking unrelated individuals following the approach described by Druet et al. [14]. However for some research purposes (milk flow trait, specific casein profiles), a few closely related individuals were sequenced and added to the overall dataset. Thus, sequence data include 13 pairs of cousins ( and 7 parent/descendant pairs (. All selected animals except 3 had previously been genotyped using the Illumina GoatSNP50 BeadChip.
A total of 2,455 French Alpine individuals (994 males, 1,461 females) and 1,570 French Saanen individuals (757 males, 813 females) genotyped using the Illumina GoatSNP50 BeadChip were available for imputation. Pedigree information was available and used as the genotyped individuals were closely related to the reference panel of sequences. Data were cleaned using an in-house pipeline as described in Martin et al. (2018). In brief, all individuals with a call rate below 95% or showing pedigree inconsistency (i.e. having more than 10% parent/offspring conflicting SNPs) were discarded. SNP quality control was based on the following inclusion criteria: call rate above 99%, MAF above 1% and Hardy-Weinberg P-value above 10-6. After editing, a total of 47,147 synthesized SNPs (out of a total of 53,347) remained on goat autosomes CHI 1 to CHI 29 and were used for subsequent analyses. Marker orders and positions were based on the ARS1 caprine Assembly [15] . The GoatSNP50 BeadChip SNP positions were updated on ARS1 genome assembly as described on the VarGoats website (http://www.goatgenome.org/projects.html) and made publically available by the International Goat Genome Consortium.
Sequence data quality check and imputation
The sequenced reads were aligned to the goat reference genome assembly ARS1 (https://www.ncbi.nlm.nih.gov/assembly/GCF_001704415.1/) using the Burrows–Wheeler Alignment tool (BWA-MEM version 0.7.15) with default parameters [16].
According to GATK best practices, BAM files were preprocessed: duplicates were removed, indels realigned and base quality score recalibrated with Picard tools version 2.1.1 and Genome Analysis Toolkit (GATK) version 3.7-0 [17]. Variant calling was performed for all GVCF files using GATK HaplotypeCaller and variants were annotated using SnpEff (version 4.3t) [18] and the NCBI Capra hircus annotation release 102 (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/001/704/415/GCF_001704415.1_ARS1/).
Variant calling on the 829 individuals led to the identification of 110,193,942 variants on the 29 autosomal chromosomes: 97,889,899 SNPs and 12,304,043 indels. Among the 829 sequenced individuals, 16 had a mean coverage below 5 (4 French Alpine and 4 French Saanen) and were removed from the data set for subsequent analyses. The sequence dataset therefore consisted of 815 sequenced individuals including 40 French Alpine goats (36 males and 4 females) and 33 French Saanen goats (31 males and 2 females). Quality checks were applied to the sequence variants using the indicators listed in Figure 1. The thresholds for individual genotype quality (GQ) and individual depth (DP) were set to 8 and 7 respectively by comparing the genotypes of the GoatSNP50 BeadChip SNPs with the sequence variants. The mean GQ and mean DP were 6.7 (± 2.8) and 7.6 (± 4.8) respectively for mismatching SNPs compared with 48.0 (± 16.7) and 11.7 (± 5.1) respectively for the matching genotypes.
After the quality check, only variants with at least one observation of the alternative allele (ALT) in French Alpine or Saanen animals were retained in order to reduce computation time in subsequent analyses and only keep variants of interest in our breeds. Thus, 23,338,436 variants, including 40,491 GoatSNP50 BeadChip SNPs, were kept for imputation. Concordance with the 50k genotypes was checked. After variant filtering, the individual mean concordance rate was 98.24% (± 1.12) and ranged from 94.00% to 99.96%.
Imputation of missing genotypes in the sequence reference panel
It should be emphasized that missing genotypes represented on average 4.63% of all the sequence variants for an individual of French Alpine and Saanen breeds. This percentage could attain 66% if sequencing was of low coverage. A within-breed imputation was therefore applied to fill in the gaps. Using a combination of AlphaImpute (v 1.9) [19] and FImpute (v 3.0) [20] gave higher concordance rates than using solely one software while minimizing computation time. We hence imputed filtered sequences using AlphaImpute and FImpute consecutively for French Alpine and Saanen breeds separately. The mean concordance rate between 50k genotypes and sequence data was 98.62% (± 1.19) after imputation and no missing genotypes remained. For subsequent analyses, as chip genotypes are more reliable than low-depth sequencing, and to avoid spreading genotyping errors down the pedigree, the 50k markers in the sequencing data were systematically replaced by information from 50k genotypes, when available.
Animal phenotypes
Male traits
Three semen production traits were recorded on artificial insemination (AI) bucks (Table 1): semen volume in mL (SV), semen concentration in billions of spermatozoa per mL (SC) and number of spermatozoa in billions of spermatozoa (SN). Semen production and quality were analyzed in 305,840 ejaculates from 2,865 AI bucks from the CapGenes breeding organization (Mignaloux-Beauvoir, France). Mean yield deviations (YD) per buck were computed from repeated performances (1 to 447 repetitions per buck) then corrected for environmental effects: age, month and year of semen collection, and time between two consecutive samples.
Female traits
Milk yield (MY) in kg was also measured as detailed by Martin et al. [6,21] and analyzed. Daughter Yield Deviations (DYD) were computed for males with at least 10 daughters with records (Table 1). DYDs were the average daughters’ performances corrected for environmental effects and merit of the dam.
Imputation scenarios of 50k genotypes to sequence level and quality assessment
Imputation of 50k genotypes to sequence level was performed using FImpute software (v 3.0) which takes pedigree information into account [20]. The accuracy and efficiency of FImpute, compared with various other imputation tools, has been confirmed [22–24]. Imputation quality was checked before imputing the available 50k genotypes to sequence level. A leave-one-out scenario was applied to 4 sequenced daughters of 2 different sequenced sires (2 Alpine and 2 Saanen) to maximize the kinship with the reference population. One of the daughters was in turn masked down to a 50k-equivalent and then imputed. The allele and genotype concordance rates (CR) and Pearson correlations (R) of the true and imputed sequences were then calculated per variant and per MAF profile.
Various reference populations were tested based on their proximity to French Alpine and Saanen goats. To accurately build the different reference panels, a Principal Component Analysis was performed on chromosome 1 filtered data using PLINK software [25]. Groups were then formed based on the origin of an individual and on the PCA results (Figure 2). The number of individuals per group is given in Table 2. All sequenced wild individuals were removed from reference populations as they are genetically different from the Capra hircus species. We also tried to impute sequences without pedigree while using all sequenced French goats available (excluding Angora and Creole breeds).
Association analysis
The imputed sequences were subjected to single-trait association analysis for milk and semen production traits using mixed linear models with the mlma option of GCTA software [26] and the following model:
See formula 1 in the supplemental files.
where y represents pre-adjusted phenotypes of the trait; µ is the overall mean; b is the additive fixed effect of the variant tested; is the vector of imputed genotypes coded in 0, 1, 2 (copy number of the alternative allele); is the vector of random additive polygenic effects, with G the genomic relationship matrix; is the vector of random residual effects normally distributed. The genomic relationship G matrix was calculated on 50k genotypes using PLINK [25].
The four traits were subjected to within-breed association analysis (Table 1). Variants with a within-breed MAF lower than 1% were excluded, leaving 11,933,965 and 12,449,740 variants in Alpine and Saanen goats, respectively, when sequences were imputed within-breed, and 14,695,413 and 15,404,361 variants in Alpine and Saanen goats, respectively, when imputation was performed using the French multi-breed panel. A Bonferroni correction was applied to the significance thresholds to account for multiple testing. The average chromosomal significance level was calculated as follows: .
The results of the sequence data association analysis were then compared with 50k-genotypes results, by performing a GWAS on the 40,491 SNPs found both in the filtered sequence data and the cleaned GoatSNP50 BeadChip SNPs.
Annotations were extracted from VCF files for variants with a -log10(p-value) above the chromosomal threshold. The RumimiR database (http://rumimir.sigenae.org/) [27] was also checked for miRNAs located close to a significant variant.
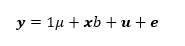{kind=link}
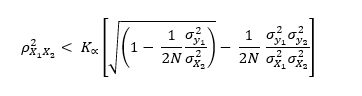{kind=link}