Experimental animals
MaleWistar rats (2 months old, 200–220g) were purchased from the animal house of Pasteur Institute (Tehran, Iran). The study was performed according to instructions approved by the Animal Care and Use Committees of Tehran University of Medical Sciences (ethical code: IR.TUMS. VCR.1398.034).Theanimals were kept in a room with controlled temperature (21–25 °C) on a 12 hours light/12 hours dark cycle.
Rat bone marrow mesenchymal stem cells (rBMMSCs) isolation and culture
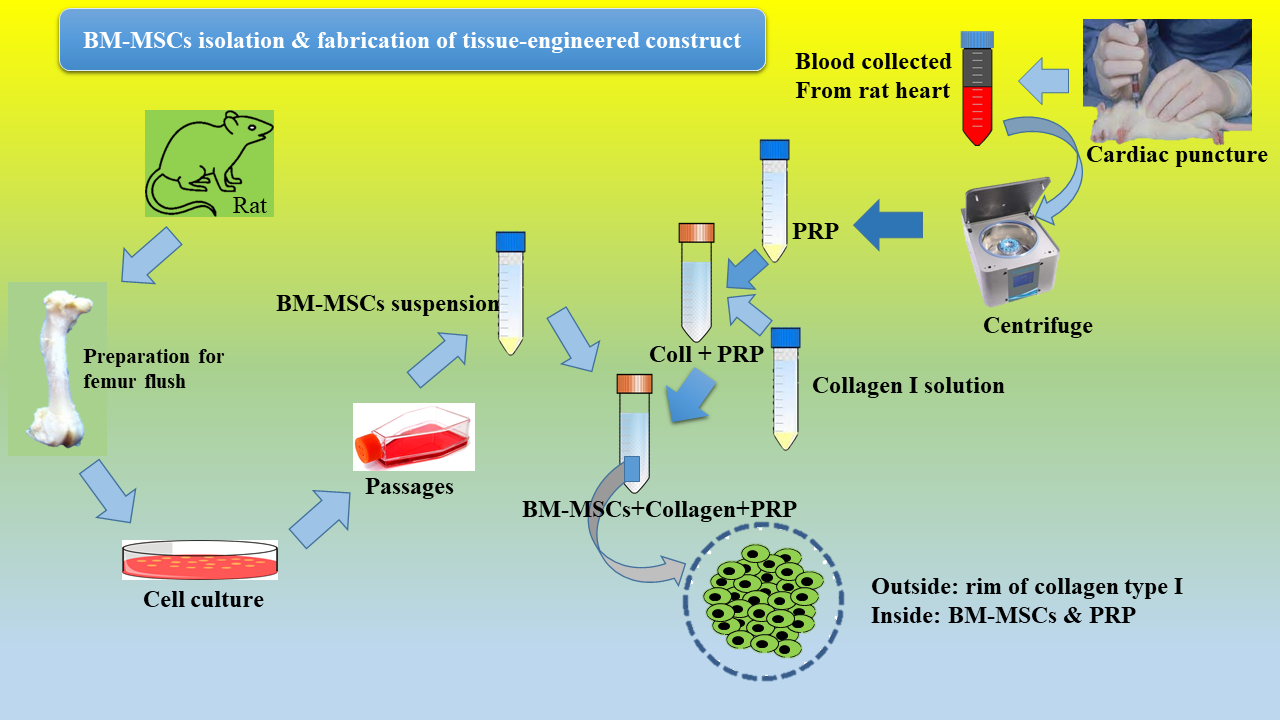
Adult rats were euthanized under proper anesthesia with ethically approved protocol. Femora and tibias were harvested aseptically. Connective tissues were removed and both ends of each bone were cut. Bone shafts were thoroughly washed with sterile PBS and the bone marrow were flushed-out with Dulbecco’s Modified Eagle’s Medium (DMEM). The cell suspension was collected in a 90 mm culture dish, then filtered by strainer (70µm) and centrifuged at 1200 rpm for 5 minand re-suspended in growth medium containing low glucose DMEM supplemented with 10% FBS, penicillin (100U/mL), L-glutamine (2mM), streptomycin (100µg/mL), and amphotericin-B (0.25µg/mL) (all from Thermo Fisher Scientific, USA). Finally, cells were plated at a density of 105 cells/cm2 in a T25tissue culture flask (Nunc, USA) and maintained at 37°C in a humidified atmosphere of 5% CO2. The medium was refreshed after 24 hours and then every 3 days. Cells at 70-80% confluency were serially passaged. We used cells at passage 3 for subsequent experiments.
rBMMSCs characterization
To characterize the cell population as multipotent MSCs, Cells at 3rd passage were characterized to meet the minimal criteria such as morphology, plastic-adherent property, expression of MSC-specific surface markers by flow cytometry and the ability for multi-lineage differentiation in vitro.
Surface marker expression
Flow cytometery(FACS Calibur, BD Bioscience, USA) was performed to determine the phenotypic expression of rBMMSCs. At 3rd passages, 1×106 cells were stained with anti-CD-90, anti-CD-105, anti-CD-45, and anti-CD-31 (All from Abcam, USA) We used the following antibodies: primary antibodiesanti-CD-105 mouse monoclonal (ab2529), anti-CD-90 mouse monoclonal (ab222781), anti-CD-34 mouse monoclonal (ab81289), and FITC-conjugated anti-CD-31 mouse monoclonal (ab33858). Unstained cells were used for iso type control. Data wereanalyzed by recording 10,000 events using FlowJo Version 7 software.
In vitro differentiation of rBMMSCs
After three passages, cells were re-plated in growth medium at 2x105cells/well in 24-well tissue culture plates. After 24 h incubation, the growth medium was replaced with osteogenic differentiation medium containing dexamethasone (10nmol), β-glycerophosphate (10mmol), L-ascorbic acid (0.3mM) (all from Sigma-Aldrich, USA). For adipogenic differentiation, 2x105rBMMSCs/well were cultured in 24-well tissue culture plates and after 24h incubation, adipogenic differentiation media containing insulin (10μL/mL), dexamethasone (1μM), indomethacin (0.5mM), and 3-isobutyl-1-methylxanine (60μM) (all from Sigma-Aldrich, USA) was added.The humidified atmosphere with 5% CO2 was maintained all through the incubation. The differentiation media was replaced every three days. After 21 days of differentiation period, the media was removed and cells were fixed and stained by alizarin red for osteoblast cells and Oil Red O staining for adipocyte cells. Calcium deposition and lipid droplets in the cells were observed using microscopy.
PRP preparation and activation
Approximately 8ml blood was collected from each euthanized rat by cardiac puncture and transferred to a centrifuge tube pre-loaded with 2ml acid citrate dextrose solution. The tubes were then centrifuged at 2000 rpm for 10 min at 20°C and plasma collected carefully then further centrifuged at 4000 rpm for 10 min at same temperature. The supernatant alone is platelet-poor-plasma (PPP) and the precipitate at the bottom of the centrifuge tube with supernatant is the PRP. When PRP volume was more than 2mL, amount 1ml of PRP was used for blood count. We activated PRP immediate prior to application with CaCl2 10% (0.2ml CaCl2 to 0.8ml PRP).
Collagen type I gel preparation and cell encapsulation
Collagen type I was extracted following the protocol described by NavneetaRajan and colleagues (25)from rat tails and processed using acetone, 70% (vol/vol) isopropanol, and 0.02N cold acetic acid. The pure acetone, isopropyl alcohol, acetic acid were purchased from Merck (Germany).The collagen solution was sterilized using 1% chloroform(Merck (Germany)) and the resultant solution was mixed with 10X DMEM-F12(Gibco, USA) and triple-buffer system-HSS at a volume ratio of 8:1:1 vol/volrespectively. The medium and HSS buffer were filtered through 0.22 µm strainer prior to mixing. The HSS buffer composed of HEPES 4.77g and 2.2g sodium bicarbonate in100 mL of 0.5M NaOH solution.All the components of HSS were purchased from Sigma Aldrich (USA). The final concentration of collagen type I hydrogel was 2mg/ml.
rBMMSCs encapsulation procedure is shown in graphical abstract. Briefly, the activated PRP and collagen type I solution were mixed properly (1:1vol/vol)in a falcontube and kept half an hour in room temperature. MSCs were harvested by using trypsin/EDTA and 2x106 cellssuspended in 100µL of complete culture media. The cell suspension was added to PRP-Collagen solution andmixed by gentlepipetting. The composition transferred and cultured in 24-well plate.
Cell morphology and attachment by SEM
The attachment of rBMMSCs in collagen was investgated by scanning electron microscope (SEM) after 48 h post cell seeding. For SEM analysis the samples were were fixed by an aqueous solution of Karnovsky’s fixative (consisting of 2.5% glutaraldehyde and 2% paraformaldehyde) for 90 min at room temperature. Thereafter, the prepared specimens were washed two times with PBS and dehydrated using a graded ethanol series of 30, 50, 70, 90, 95, and 100%. Finally, the specimens were dried under vacuum and after coating with gold, examined via SEM (SEM, AIS2300C SEI, Korea) at 20 kV.
MTT assay
MTT assay was performed to measure the viability of encapsulated cells alone and with PRP at different incubation period (day 1, 3, and 5) in a 96-well plate. This method is based on the reduction of the yellow MTT-salt [3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium-bromide] to blue-purple formazan crystals by mitochondrial enzyme succinate dehydrogenase. For this purpose, 1×104 cells (encapsulated) were plated on each well with PRP (controls without PRP). Then 200μL MTT solution (5 mg/mL) was added in each well andthe plate was incubated at 37°C for 3–4 hours. Then the supernatant was replaced with 100μL DMSO in order to dissolve formazan crystal producing purple-blue colour. Optical density measured by using microplate readerat 570nm.
In vitro chondrogenic differentiation of encapsulated rBMMSCs
After passage 3, the cells, at a concentration of 2x106 cells/ml, encapsulated in collagen solution then cultured in the 24-well tissue culture plate. After 4h incubation, differentiation groups were divided to three groups. 1. TGF-β1group: treated with chondrogenic differentiation media that contains DMEM/F12 supplemented with 10%ITS+ 10−7 M dexamethasone (Sigma), 1μM ascorbate-2-phosphate, and 10ng/ml transforming growth factor-beta 1 (TGF-β1, SIGMA,USA) 2. PRP- TGF-β1 group: treated with chondrogenic differentiation media +PRP with a ratio of 1:1, and 3. Control group without differentiation media and PRP. The cell culture maintained for 21 days with media change in every 3 days. Finally, the cells were collected for the real time PCR and were fixed for immunocytochemistry for expression of related genes to chondrocyte.
Real time PCR
Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) was used for the mRNA expression patterns of chondrogenic specific genes in control, TGF-β1 and PRP-TGF-β1 groups. Total RNA was prepared by using RNeasy Plus Mini kit (Qiagen, USA, 74134) as described by the manufacturer, and complementary DNA (cDNA) synthesis from 1μg of extracted RNA was performed by Revert Aid First Strand cDNA Synthesis kit (Takara, USA, K1632). qRT-PCR reactions were carried out in the 48-well optical reaction plates on StepOneTM Real-Time PCR machine. For each PCR reaction, 30ng synthesized cDNA was used for PCR by mixing with 10μl of Power SYBER Green master mix (2×, Applied Biosystems) primed with 0.5 μM of each primer (Table 1) in a total volume of 20μl at the annealing temperature. The comparative Ct method (ΔΔCT Method) was used for relative gene expression analysis. All Ct values calculated from the normalization of target genes to GAPDH as an internal control and calibrated using calculation from the undifferentiated BM-MSCs. The relative gene expression values presented as mean of three independent experiments.
Immunocytochemistry
After 21 days' post-induction to chondrogenic differentiation, encapsulated cells were fixed with 4% paraformaldehyde (PFA; Sigma-Aldrich) for 20 minutes. Subsequently permeabilized with 0.1 % Triton X-100 in PBS. The cells were blocked for 30 min at room temperature with 5 % bovine serum albumin (BSA), thenincubated with primary antibodies (diluted in 5 % BSAin PBS)against aggrecan (mouse monoclonal antibody; Abcam, USA, 1:200), collagen type I (mouse monoclonal antibody; Abcam, 1:200), and collagen type II (mouse monoclonal antibody; Abcam,1:200) overnightat 4°C. Secondary antibodies included Alexa fluor 488 donkey anti-mouse (1:500; Gibco) and the nuclei were counter-stained with DAPI (Sigma-Aldrich, D8417). For negative controls, only the secondary antibody was used.
In vivo experiment
The 8 weeks male wistar rats (200-220g) were randomized into four groups each of 6 rats: (i) OA group (OA induction without treatment), (ii) OA+MSC group, (iii) OA+Collagen+MSC+PRP group, and (iv) OA+MSC+Collagen group. OA of the knee joints was induced as previously described by Tang et al (2017). In brief, under sterile conditions, the animals were anesthetized and a medial parapatellar approach was used to release the joint capsule and to perform a medial collateral ligament transection (MCLT) of the right knee joints. The wounds were closed and covered with a local antibiotic ointment. All rats were returned to their cages after the operation and were allowed to move freely, and 0.2 mg/kg/day of intramuscular Meloxicam was administered for 5 days for pain relief and administrated by penicillin once a day for the first 3 days. On fourth weeks post-induction,OA was confirmedbyradiography.Then the knee joints were injected by MSC, Collagen+MSC, and Collagen+MSC+PRP. The recipient rats were injected with 4X106 collagen-encapsulated MSCs (passage 3). All rats were euthanized 6 weeks post-treatment, and knee joint samples were collected for histopathological evaluations.
Statistical analysis
Tukey's HSD test and repeated measures analysis of variance test was used for multigroup comparisons according to the GraphPad Prism software, Version6.00 (GraphPad Prism, Inc., San Diego, CA). P<0.05 was considered significant.
{kind=link}