We analyzed the repertoire of genes expressed in cortical microglia from both control and AD-mice (Figure 1A) by combining laser capture microdissection (LCM) and RNA-seq approaches in transgenic mice expressing eGFP under the control of the CX3CR1 promotor [24, 25]. Moderate tissue fixation preserves fluorescence but alters the quantity and quality of recovered RNA, whereas eGFP fluorescence is generally low in fresh unfixed brain samples [38] (and Data not shown). To overcome these issues, we developed a specific protocol based on tissue preservation by sucrose perfusion and immersion, rapid freezing in -40°C isopentane, cryo-sectioning and dehydration, which allows eGFP fluorescence preservation, amyloid plaque staining as well recovery of RNA in good quality and quantity (Figure S1A).
We extracted total RNA from 400-600 microglia per animal per experimental condition (Table S1) and performed mRNA sequencing. We detected 13,923 expressed genes, and compared the expression of 133 randomly selected ones (i.e. exhibiting low, medium or high expression levels) in FACs sorted microglia ([33]). Linear regression revealed a significant correlation between gene expression levels in LCM and FACS isolated microglia (r=0.697, p<0.0001), demonstrating that our data are consistent with previously published microglia gene expression profiles (Figure S1B).
Isolation of microglia from mouse brain tissue with preservation of spatial information
Cell isolation through LCM allows preservation of spatial information, but is subject to cross-contamination by surrounding cells whose processes may by be captured together with the cell of interest. To assess the degree of microglial enrichment, we evaluated the expression levels of specific microglial, astrocytic, oligodendrocytic and neuronal genes in the different LCM samples. Figure 1B shows that microglia specific genes are about 10 times enriched in the LCM samples compared to the whole cortex tissue, whereas, reversely, other glial cells and neurons specific genes are strongly depleted. In CX3CR1+/eGFP mice, eGFP is expressed in all myeloid cells including infiltrating monocytes that can penetrate brain parenchyma in pathological conditions [39]. To assess the possibility that our LCM samples could be contaminated by infiltrating monocytes, we analyzed the expression of peripheral monocyte/macrophage specific (Figure 1B). Interestingly, we demonstrated that the expressions of Cd163 and S100a4 were also depleted in the LCM samples. Cd74 expression was depleted in control microglia (CM), but enriched in PAM and PCM indicating that this gene is upregulated in reactive microglia.
An important drawback in cell isolation is the intrinsic cell activation induced by generating single-cell suspension. This is particularly true for microglia which are inherently reactive cells, for which it has been shown that FACS sorting may alter the analysis of the disease-induced transcriptomic changes [19, 40]. In contrast to FACS, in LCM, cells are isolated from their environment after dehydration, which prevents the cell reaction as shown by low expression of immediate early genes in the LCM isolated microglia (Figure 1C).
The reproducibility of our data was demonstrated by the strong correlation between the biological replicates (Figure S1C). In addition, principal component analysis on mRNA expression profiles allowed good discrimination between the different microglia subtypes (Figure 1D). In particular, PAM clearly separate from both CM and PCM. Interestingly, PCM also segregate from CM. However, this analysis did not allow further separation of the samples by age within each sub-population. We then examined, in the different subpopulations, gene expression for markers of previously identified microglia gene signatures, namely markers of homeostatic microglia (P2ry12 & Tmem119, Figure 2A), Disease-associated microglia (DAM, Apoe & Ctsd, Figure 2B, [11]), Activated-response microglia (Cst7 & H2-ab1, ARM, Figure 2C, [13]) and Interferon-response microglia (Ifit2 & Ifit3, IRM, Figure 2D, [13]). Our results showed that homeostatic microglial gene expressions remained stable in CM and PCM, and only appeared to decrease in late stage PAM. Interestingly, although DAM, ARM and IRM markers are expressed at higher levels in PAM, our results revealed that these genes are also up-regulated in PCM with their expression increasing in an age-dependent manner. Notably, expression of DAM and ARM markers remained stable across disease stages in PAM, whereas that of IRM appeared to increase in older PAM (Figure 2C-D).
Having established that this protocol allows the isolation of spatially distinct sub-populations of microglia with minimal intrinsic perturbation and sufficient enrichment, we conducted specific contrast analyses to identify DEGs. We then performed bioinformatic/biostatistic analyses to infer the pathophysiological role of the different microglia subpopulations in AD progression. The workflow for data analysis is presented in Figure S2.
Biological functions and Master gene regulators in PAM
In the APP/PS1 model, dense amyloid plaques begin to appear in cortical areas at about 4-months of age [25]. However, at this age, plaques are very sparse and it was not technically feasible to isolate PAM. Thus, to identify gene deregulation in PAM versus CM microglia, we restricted the analysis to 8-mo and 12-mo samples.
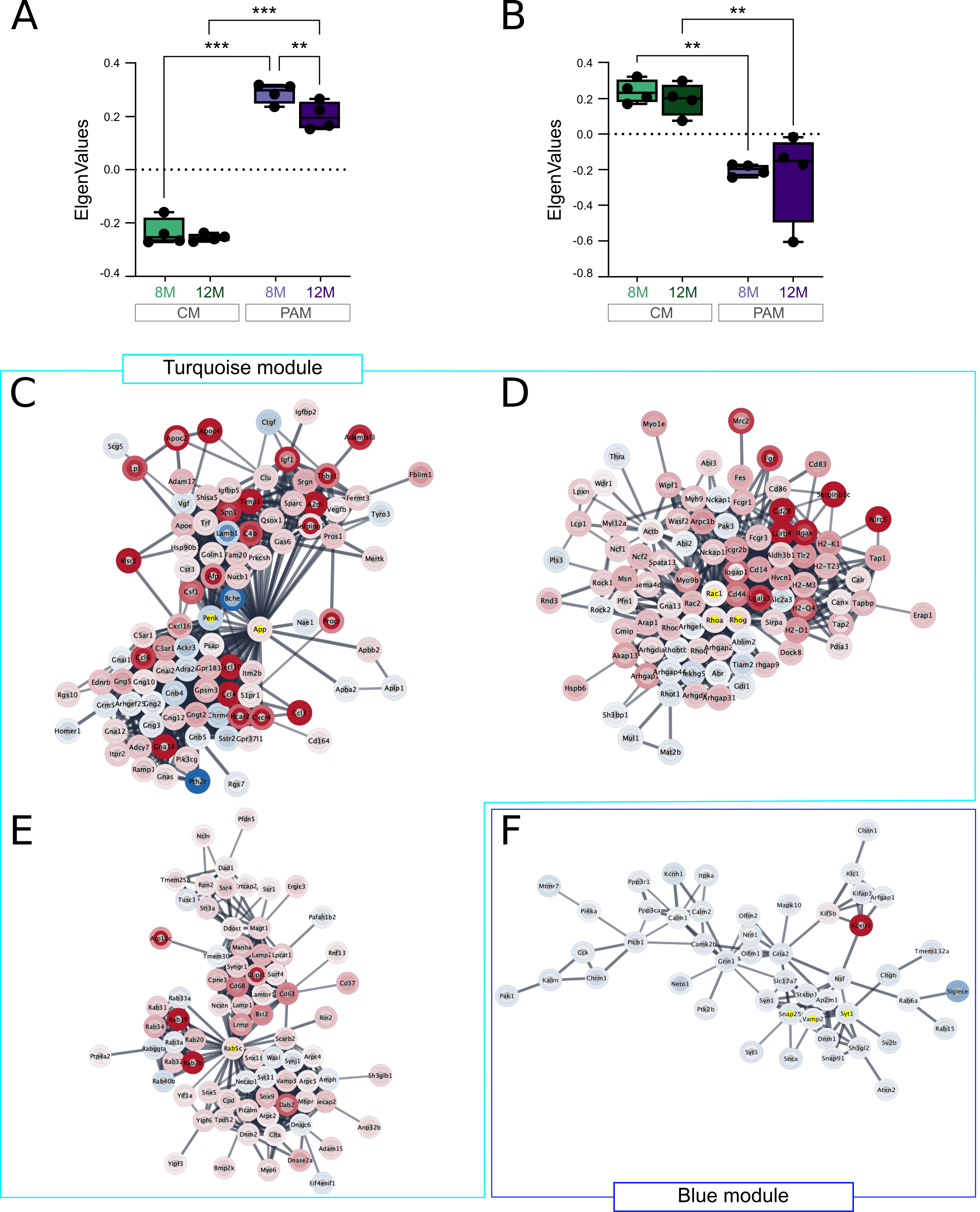
PCA revealed a clear distinction between CM and PAM, and statistical analysis identified 1851 DEGs (false discovery rate (FDR) <0.05), about two-thirds of which were up-regulated (Figure 3A and data not-shown). Previous studies established microglia reaction signatures in different pathological conditions. This includes the DAM, ARM and IRM signatures previously mentioned [11, 13], but also signatures for Inflammatory Associated Microglia (IAM) which represent a large set of DEGs in inflammatory conditions [34] and for the Reactome a smaller set of 86 genes deregulated in different acute and neurodegenerative conditions [33]. Figure 3B shows that, compared to control microglia, PAM DEGs were significantly enriched for these different pathological microglia gene signatures. The Sensome gene signature represents a set of membrane-associated proteins and receptors that are selectively expressed in microglia and that help them sense changes in their environment [32] (refined in [33]). This signature was also significantly affected in PAM. Using a spatial transcriptomic approach, Chen et al. recently identified a plaque-induced gene (PIG) network mainly involving microglial and astroglial genes [20]. As expected, PAM signature was strongly enriched with PIGs genes, actually 52 of the 57 PIGs genes were deregulated in PAM (Figure 3B).
Genes with similar expression patterns (co-expressions) are likely to have similar functions and can be grouped into modules by WGCNA [30]. We performed WGCNA analysis to identify gene modules among the DEGs, and GO based enrichment analyses to extract the hypothetical biological functions for each of these modules (see Figure S2 and Materials and methods for details). Among the 1851 DEGs, we identified two distinct modules significantly correlated with the Microglia-subtype trait. The largest module (i.e. turquoise module) included 1639 genes mainly up-regulated in PAM (Figure S3A and Table S2A). These genes were primarily associated with inflammation related biological processes, including Cell activation & proliferation, Immune response, Cytotoxicity, Exocytosis, Chemotaxism, Antigen presentation, etc. (Figure 3C; Table S2B). Alterations in Cell morphology was another significantly affected biological function. The second module (i.e. blue module) was smaller and contained 212 genes mainly down-regulated in PAM (Figure S3B; Table S2A). This module mainly related to Synaptic transmission associated biological processes (Figure 3D; Table S2C).
In gene networks or subnetworks, hub genes (i.e. most highly connected genes) represent master regulators that are likely to play essential roles in controlling the biological response. We used specific applications in Cytoscape (see materials and methods) to first construct the genes’ network of the two WGCNA modules, and second to identify the most connected sub-networks and their potential hub genes. In the Turquoise module, the 1074 most highly connected genes were separated into subnetworks using the MCC cluster tool. The ten larger clusters are detailed in Table S3, with hub genes highlighted in dark green. The three larger subnetworks are also shown in Figure S3B-D, with hub genes in yellow. In the largest subnetwork, App and Penk which are respectively up- and down-regulated in PAM appeared to play orchestrating and redundant roles for controlling chemotaxis and endopeptidase activities (Figure S3B). The second subnetwork included genes that control cell shape and antigen processing. Hub genes of this network were the GTPases Rac1, Rhoa and Rhog which belong to, respectively, the Ras and Rho super-families (Figure S3C). Rab5c is another small GTPase involved in controlling receptors endocytosis, vesicle trafficking, and endo-lysosomal pathways [41] and which played a central role in the third largest subnetwork (Figure S3D). In the blue module, we identified a single network of 48 highly connected genes (Figure S3F). This gene network was associated with control of the synaptic vesicle cycle (Table S3); hub genes were Syt1, Vamp2 and Snap25 which represent key proteins for neurotransmitter release.
Next, we addressed the question of the extent to which age affected the transcriptomic changes observed in PAM. To meet this goal, RNA-seq data from the 8-mo and 12-mo samples were reanalyzed using a Generalized Linear Model (GLM) model, with Microglia-subtype (PAM vs CM) and Age (8-mo vs 12-mo) as between samples’ factors (Figure S2). Thus, we identified 723 DEG in the Microglia-subtype:Age interaction (raw p-value<0.05). Among them, we restricted our analysis to the 179 genes that were significantly deregulated in PAM vs CM (Figure S4A). WGCNA analysis further identified three genes modules (turquoise, blue and brown) that were significantly correlated with the Microglia-subtype trait. The largest module (i.e. turquoise module) included 96 genes that related to (1) Wound healing, Cell projection organization and PKB signaling biological processes; (2) Bacterial invasion and Phagosome KEGG pathways and (3) Ephrin signaling KEGG pathways (Figure S4C). Interestingly, these genes were more strongly over-expressed in 8-mo versus 12-mo PAM and were slightly over-expressed in 12-mo CM suggesting that the normally occurring overexpression of these genes during normal ageing was accelerated in PAM (Figure S4B). The blue module regrouped genes that were mostly down-regulated in PAM and tended to be less expressed / more down-regulated in 8-mo PAM. Moreover, these genes were also down-regulated in 12-mo CM. Genes of this module did not relate to any specific GO biological processes, but were associated with MAP kinases, ErbB signaling and mitophagy KEGG pathways (Figure S4C). Finally, genes of the brown module are upregulated in PAM but down-regulated in older CM (Figure S4B). These genes are associated to hypoxia related and cell adhesion pathways (Figure S4C).
Overall, our comparison of PAM versus CM in the APP/PS1 model shows that PAM exhibit profound transcriptomic changes which drive an increased inflammatory reaction, support morphological changes and contribute to the degradation of synaptic support functions. Although location at the proximity of Ab-plaques is the most important driver for transcriptomic changes, Age contributes, but to a lesser extent, to the observed alterations.
Biological functions and master gene regulators in PCM
Because amyloid plaques relate to one of the most prominent features of the disease, studies on the role of microglia in Alzheimer’s disease have often focused on PAM. Quite the reverse, PCM whose morphology is very similar to that of CM are generally overlooked (Figure S5A). However, these cells are also part of the pathological environment and we reasoned that they are likely to also contribute to the disease progression.
To investigate whether specific biological functions were altered in PCM versus control CM, we identified genes significantly deregulated between the two conditions irrespective of age. PCA discriminated PCM from CM according to the second dimension (Figure S5B), and statistical analysis identified 102 DEGs (FDR <0.05), the great majority (87/102) of which were up-regulated (Figure 4A, Table S4A). Interestingly, as for PAM, deregulated PCM DEGs were very significantly enriched for the different pathological microglia gene signatures (i.e. IAM, DAM, ARM, MGnD, IRM signatures), the Reactome and the Sensome signatures (Figure 4B). More surprisingly, PCM’s DEGs were also highly enriched for PIG gene network. However, this may be explained by the fact that in Chen et al. [20] study, amyloid load was quantified based on 6E10 immunostaining which labels more diffuse Ab plaques. GO analyses also revealed that these genes are associated with immune related functions, including Tumor Necrosis Factor (TNF) and Cytokine production, Immune response and Antigen presentation (Figure 4C; Table S4B). Cellular reaction in this microglia subtype was also demonstrated by deregulation of functions linked to Cell differentiation and Myeloid activation. Among those DEGs, WGCNA analysis identified 2 distinct modules of co-deregulated genes (Table S4A). The largest one contained the vast majority of the DEGs (91/102) and corresponded to genes that showed an age-dependent upregulation in PCM (Figure 4D). The second module was limited to only 7 genes, including App, which by construction is over-expressed in the APP/PS1, and could not be related to a specific biological function (data not shown). On the other hand, gene network analysis identified a cluster of 49 highly connected genes that are strongly associated with the Lysosome (p= 4.1x10‑11), the Antigen processing & presentation (p= 2.4x10-10), and the Phagosome (p= 7.4x10-6) KEGG pathways (Figure 4E). Cd68, Ctsd, H2-aa and C3ar1 represented hub genes within this network.
As shown in Figure 4A, gene expression changes were quite variable in PCM with a general trend for higher deregulation in microglia isolated from older mice. Additionally, although the expression changes were more similar within 4-mo and 12-mo samples, the inter-individual variation appeared greater in microglia isolated at in 8-mo mice. To address whether age affected the transcriptomic changes observed in PCM, we first identified 1334 genes deregulated in PCM versus CM (raw p-value<0.05), and then searched among them which ones are also deregulated in the Microglia-subtype:Age interaction (raw p-value<0.05) (Figure S5C). We thus identified 595 genes whose expression changed in PCM in an age-depend manner. WGCNA analysis further identified two gene modules. The largest one (turquoise module) was significantly correlated with the Microglia-subtype factor. Genes of this module were up-regulated in the intermediate and late stage of the disease (Figure S5D, upper panel) and related to inflammatory processes, notably Cytokine production, Antigen presentation, Myeloid cell activation and the Phagosome KEGG pathway (Figure S5E). Interestingly, these genes showed opposite regulation in CM being less expressed in 12-mo compared to younger cells. The second module (blue module) was significantly correlated with the Age factor and contained genes whose expression were down-regulated in an age-dependent manner specifically in PCM (Figure S5D, lower panel). Genes of the blue module related to Lipid oxidation, Organelle transport and Synaptic transmission biological processes (Figure S5E).
On the whole, these results demonstrated that although PCM are not associated to Ab plaques, and display homeostatic-like morphology, they exhibit age-dependent transcriptome alterations. These alterations are associated with important microglial functions that are typical of microglial reaction.
To what extent do PAM and PCM differ?
To further investigate the extent to which PAM and PCM differ at the transcriptomic level, we searched for genes significantly deregulated between the two microglia subtypes. Considering both the 8-mo and 12-mo samples, we identified 551 DEGs (FDR < 0.05), of which 80% (446/551) were up-regulated in both PAM vs PCM (Figure 5A). WGCNA analysis identified a single module of 497 genes, which was significantly correlated with the Microglia-subtype trait (r=0.95; p<2.10-8) and more highly expressed in PAM (Figure 5B, Table S5A). These 497 genes were associated with inflammation related biological processes (Figure 5C; Table S5B) and, at least in part, overlapped with those deregulated in PCM vs CM, indicating that PCM present an intermediate reactive state between CM and PAM. However, some biological functions were specific to PAM, including Chemotaxism, Cell proliferation, Cell architecture and ROS production. By comparing the three lists of DEGs (i.e. PAM vs CM, PCM vs CM and PAM vs PCM), we also identified 11 genes that were deregulated in PCM only (Figure 5D). Globally, these genes showed significantly greater expression in PCM compared to CM, whereas in PAM their expression was either not different or lower (i.e. 1 gene, Efnb3) than in controls (Table S6). This latter result suggests that PCM are also engaged in specific functions compared to PAM. However, these small panel of genes could not be associated to any specific biological processes (not shown).
Among the genes deregulated in PAM versus PCM, we then identified 96 genes that were changed in Microglia-subtype:Age interaction (raw p-value<0.05) (Figure S6A). WGCNA analysis refined this list to 91 co-expressed genes that were more highly expressed in PAM compared to PCM. These genes were enriched for biological processes associated with Actin filament organization, Cell migration & differentiation, Peptidase activity (Figure S6B and Table S7) and for the KEGG Chemokine signaling pathway (Table S7). They showed opposite age‑dependent regulation in PCM and PAM, and thus globally appeared less up-regulated in 12-mo PAM (Figure S6C and Figure S6D).
As a whole, these results indicate that although PAM and PCM share common signaling pathways, they are also engaged in specific biological functions. Our data also reveal different age-dependent regulations in PCM and PAM.
To further explore the relative contribution of PCM and PAM to AD, we tested whether AD risk genes were enriched among the PAM and PCM DEGs. To that purpose, we used a list of genes from a recent and extensive GWAS study, converting the human ID genes for their murine orthologs [42]. Recent studies on polygenic risk scores have shown that genes with even small significance in GWAS carry information with regard to the risk of AD [43], thus we tested for enrichment in GWAS genes at different cutoffs (Figure 5E). At all cutoffs (p-values ranging from 10-6 to 10-2), PAM DEGs were significantly enriched for GWAS-associated AD genes thus suggesting that PAM play a key role in AD pathogenesis. In contrast, PCM DEGs were significantly enriched for GWAS AD genes only at the lowest cutoffs. This suggests that PCM can contribute to AD pathogenesis, although, to a lower extent than PAM.
Validation in brain tissue
Laser microdissection can be used to isolate discrete cells from complex environments while preserving spatial information, however, the cell populations obtained by this method are not pure. To validate the cellular and spatial localization of the DEGs, we selected 3 genes (i.e. Cst7, Cybb and Clec7a) that show significant deregulation in both PAM and PCM and performed single-molecule fluorescence in situ hybridization (smFISH) with specific RNAScope probes against these targets (Figure 6 & S7-S8). smFISH was coupled with immunofluorescent detection of microglia using anti-GFP antibody and amyloid plaques using ThiazinRed staining (Figure 6 and Figure S7). In agreement with the robust up-regulation of their mRNAs in PAM, Cst7, Clec7a and Cybb signals strongly colocalized in microglia associated with amyloid plaques (Figures 6A,C [Cst7] ; S7A,C [Cybb] and S8A,C [Clec7a]). However, weaker but positive RNAscope signals for Cst7, Clec7a and Cybb were also observed in microglia located further than 70 µm away from plaques (Figures 6A,D [Cst7] ; S7A,D [Cybb] and S8A,D [Clec7a]). We also observed a pronounced heterogeneity in the level of expression of these mRNAs in microglia, whether or not they were associated to Ab plaques. Indeed, some microglia expressed high levels of target mRNA, while neighboring microglia expressed little or no mRNA at all (Figure 6C; S7C and S8C; see arrow-heads).
{kind=link}