Recent genomic studies of parasitic plants have revealed that there are numerous footprints indicative of horizontal gene transfer (HGT) to the parasites from their host plants. However, the molecular mechanisms and biological impacts of this phenomenon have remained largely unknown. Here, we made the striking observation that two parasitic dodders, Cuscuta campestris and C. australis, have functional homologues of Si_CYP81Q1, which encodes piperitol/sesamin synthase (PSS) in the phylogenetically remote plant Sesamum indicum (sesame). The apparent lack of sequence similarity between the regions flanking PSS in Sesamum and Cuscuta spp. suggests the occurrence of HGT tightly associated with the PSS gene. Upon parasitism, C. campestris induced expression of the host Si_CYP81Q1 at the parasitic interface and mature and intron-retained Si_CYP81Q1 mRNA was transferred to C. campestris, suggesting that CYP81Q1 was translocated via RNA-mediated HGT. Thus, parasitism-evoked HGT might have had an unexpected role in the metabolic evolution of plants.
Research Article
Parasitism-evoked horizontal gene transfer between plants as a novel trigger for specialized metabolism evolution
https://doi.org/10.21203/rs.3.rs-885568/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version
Cuscuta
Horizontal gene transfer
Metabolic evolution
Parasitism
Sesumum
Cuscuta spp. (Convolvulaceae, Solanales), commonly known as dodders, are obligate parasitic plants with a broad host range1,2. In a recent genome analysis, one species, C. australis, was estimated to be separated by about 55 MYA from the phylogenetically related morning glory (Ipomoea nil., Convolvulaceae, Solanales)3. Cuscuta plants are rootless and leafless, and thus have limited or no photosynthetic ability4. To obtain biochemical resources for survival and proliferation, Cuscuta have acquired the ability to form connections with autotrophic host plants through a specialized root-like structure called the haustorium, which enables acquisition of sugars and minerals from the host. Notably, recent studies have shown that Cuscuta plants also take in genomic DNA and messenger RNA from host plants5,6. Interestingly, the strictosidine synthase-like gene in C. australis showed a much higher sequence similarity with related genes in Brassicaceae than genes in close relatives of C. australis, suggesting HGT from Brassicaceae host plants to the parasites7. C. australis also acquired an acyltransferase gene from Fabaceae host plants8. These data suggest that the Cuscuta genome has acquired a substantial number of genes during its evolution; however, whether such HGT-derived genes in Cuscuta are functional has not been clarified, and the mechanisms by which genetic molecules are delivered from host plants to Cuscuta have not been elucidated.
Despite their lack of photosynthetic ability, Cuscuta plants are known to produce a variety of bioactive compounds. Cuscuta spp. are used in Asian traditional herbal medicine for anti-aging, anti-inflammatory, hepatoprotective, pain reliever, and aphrodisiac purposes9,10. Previous studies demonstrated that Cuscuta spp. contain a variety of specialized bioactive metabolites including flavonoids, steroids, and alkaloids10,11, which might represent adaptive metabolic evolution to specific situations and might reflect the unique physiology of these plants.
Specialized metabolites are lineage-specific and usually restricted to a phylogenetically close group, known as a chemotaxonomic group, due to the common biosynthetic origin of an ancestor e.g., isoflavonoid in Fabaceae. However, some specialized metabolites are sporadically found in phylogenetically unrelated plants, forming metabolic ‘patchiness’ in chemotaxonomy. The sporadic occurrence of the specialized metabolite can be explained by convergent evolution (CEV) based on the independent occurrence of biosynthetic genes, which produce common metabolites in several plants. For example, a specialized metabolite derived from xanthine, caffeine, can be found not only in Camellia sinensis (tea) and Coffea arabica (coffee), which are both in the Asterid group, but also in Theobroma cacao (cacao) and Citrus sinensis (orange), which are in a distinct clade, the Rosids. The discontinuous presence of caffeine has been explained as CEV of caffeine synthase genes12. More recently, CEV of a biosynthetic gene cluster for momilactone biosynthesis was uncovered in different plant lineages13, indicating that CEV is the genetic basis for sporadically distributed metabolites in plants.
Sesamin is another example of a sporadically occurring specialized metabolite. Lignans are a large class of phenylpropanoid-dimeric metabolites found in plants. Sesamin is the major lignan in seeds of Sesamum spp. (Pedaliaceae, Lamiales) and is also found in phylogenetic relatives of Sesamum, e.g. Pedaliaceae and Paulowniaceae14. In sesame seeds, a cytochrome P450 monooxygenase of S. indicum, Si_CYP81Q1, also known as piperitol/sesamin synthase (PSS), forms two methylenedioxy bridges (MDB) on the two aromatic rings in the sequential conversion of pinoresinol to piperitol and then to sesamin15. The strikingly homologous CYP81Q genes are functionally conserved in phylogenetic relatives of S. indicum, which produce sesamin and its related metabolites15,16,17. This is an example of a typical metabolic radiation within a restricted taxonomic group sharing a common biosynthetic gene.
In addition to being found in Lamiales, sesamin is also discontinuously observed in phylogenetically unrelated plants such as Piper spp. (black pepper), Magnolia spp. (a basal angiosperm), and Ginkgo (Gymnosperm)14,18,19. Given the lineage specificity of specialized metabolites, it is remarkable that Cuscuta spp. commonly contain sesamin and related metabolites such as cuscutoside (Supplementary Fig. 1), since thus far, sesamin has not been reported in Convolvulaceae other than Cuscuta9,14,20,21,22. Notably, sesamin is detected in Cuscuta palaestina, which is a parasite of host plants that do not accumulate sesamin, suggesting that C. palaestina de novo produce sesamin in planta rather than absorbing it from host plants22. Thus, sesamin serves as an excellent example of the sporadic occurrence of a specialized metabolite. Nevertheless, how Cuscuta acquired the ability to produce sesamin has not yet been elucidated (Fig. 1a).
The PSS genes in Lamiales are the only genes known to encode proteins with sesamin synthase activity. There are at least three possible explanations for the sporadic occurrence of sesamin: 1) functional differentiation or gene loss (FD/GL), 2) CEV of a sesamin synthase, and 3) HGT of a sesamin synthase gene. A previous report on FD of sesamin synthase showed that S. alatum CYP81Q3 produces pluviatilol instead of sesamin, resulting in the exceptional absence of sesamin in S. alatum among the Sesamum spp.17. FD/GL is likely restricted to a relatively small taxonomic group due to its dependence on metabolic radiation coupled with a common biosynthetic gene. By contrast, CEV occurs in distant, unrelated phylogenies, free from metabolic radiation. As a result, CEV genes tend not to be similar due to their distinct genetic origins. HGT provides an alternative explanation for the presence of sporadic metabolites and likely occurs in parasitic plants such as Cuscuta, Striga, and Orobanche spp. via the unusual physical interaction these plants have with their hosts. In this case, HGT genes are expected to be structurally similar to genes originally encoded in host plants. Identification of sesamin synthase genes from non-Lamiales plants would not only clarify the metabolic origins of sesamin but also shine new light on the nature of metabolic evolution of specialized metabolites.
In this study, we attempt to unravel the origin in Cuscuta plants of sesamin and structurally related metabolites with MDB by integrating comparative genomics, parasitism testing, and biochemical approaches. The results obtained using these different approaches support the unexpected finding that parasitism-mediated functional HGT is a possible driving force for sporadic metabolic evolution.
Identification of Cuscuta CYP81Q-related genes
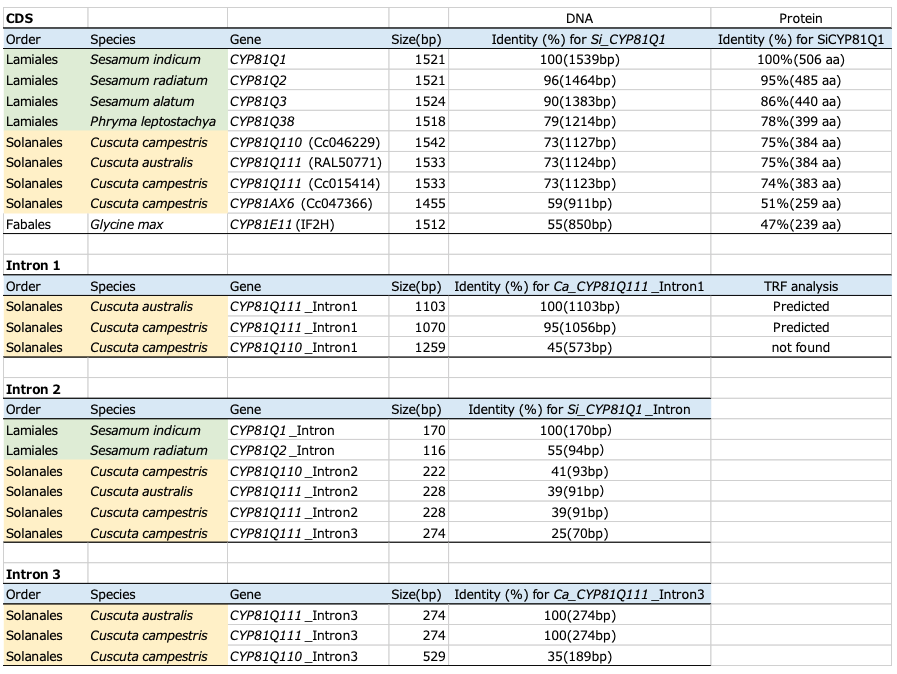
We analyzed lignans in C. campestris seeds and detected sesamin and related lignans (Supplementary Fig. 1). To elucidate the molecular basis of sesamin biosynthesis in Cuscuta plants, we investigated genome and transcriptome data of C. campestris by Basic Local Alignment Search Tool (BLAST) search using Si_CYP81Q1 (AB194714) as a query23. We found two CYP81Q1 homologs, Cc_CYP81Q110 (Cc046292) in scaffold18 and Cc_CYP81Q111 (Cc015414) in scaffold84. Both of these genes are predicted to have three introns, their putative amino acid sequences are 95% identical, and they share ca. 75% identity with Si_CYP81Q1 (Fig. 1b, Table 1, Supplementary Table 1). We identified Cc_CYP81AX6 (Cc047366) as a third homolog of CYP81Q1 in the C. campestris genome; however, Cc_CYP81AX6 has relatively low amino acid identity to Si_CYP81Q1 (51%) and formed a distinct phylogenetic cluster together with isoflavone hydroxylases from Fabaceae such as Ge_CYP81E124,25 and CYP81E-related genes from I. nil. This cluster is distinct from that harboring Cc_CYP81Q110 and Cc_CYP81Q111. By contrast, no genes with striking similarity to Si_CYP81Q1 were observed in the I. nil genome or transcriptome26.
We also found a single P450 gene, Ca_CYP81Q111 (C065N002E0.1), that has high structural similarity to Si_CYP81Q1 (75% amino acid identity) in another (+)-sesamin producing dodder, Cuscuta australis, which is phylogenetically related to C. campestris and whose genome has been sequenced (Fig. 1b, Table 1, Supplementary Table 1)3,14. The size of the C. australis genome (c.a. 264 Mbp) is approximately half of that of C. campestris (c.a. 556 Mbp). This is consistent with the idea that the C. campestris genome doubled in size due to either a recent whole genome duplication (WGD), estimated to have occurred in the C. campestris genome around 1.5 MYA after its divergence from C. australis or via a hybridization event between phylogenetic relatives2,3,21,23. Based on this, we concluded that Ca_CYP81Q111 is singlet in the genome, whereas Cc_CYP81Q110 and Cc_CYP81Q111 are twin homeologs.
We next used a bioinformatics approach to try to construct the DNA sequences of Cuscuta CYP81Q-related genes using public NGS data of the following Cuscuta plants: C. californica, C. gronovii, and C. americana from subgenus Grammica; C. europaea and C. epithymum from subgenus Cuscuta; and C. reflexa, C. japonica, and C. monogyna from subgenus Monogynella, which is the most primitive group of Cuscuta and is similar to nonparasitic relatives1. Partial sequences including exon 2 of Cuscuta CYP81Q genes were found by BLAST search in Grammica and Cuscuta, but not in the three species of Monogynella (Supplementary Fig. 2a). The absence of CYP81Q-related genes was experimentally examined by PCR using primers designed for exon 2. No amplification was observed from genomic DNA of C. japonica belonging to Monogynella as a template while a specific band was amplified from that of C. campestris belonging to Grammica (Supplementary Fig. 2b). Additionally, sesamin was not detected in seeds of C. japonica (Supplementary Fig. 1), consistent with the idea that there is no CYP81Q-related gene in this species. The three exon 2 sequences from subgenus Grammica clustered with those of C. australis and C. campestris, whereas the two sequences of subgenus Cuscuta formed a phylogenetic clade distinct from that of Grammica, roughly in line with the phylogeny of Cuscuta spp.
These results support the notions that 1) the CYP81Q-related gene is widely conserved among subgenera Grammica and Cuscuta, 2) the origin of this gene likely ascends to a monophyletic ancestor located between subgenera Monogynella and Cuscuta, and 3) the gene has undergone nucleotide diversification from the common ancestral CYP81Q orthologue along with speciation of the subgenera Cuscuta and Grammica.
In contrast to Cuscuta spp., we did not find any homologs of Si_CYP81Q1 in the public genomes of Ginkgo biloba, Magnolia ashei, or Piper nigrum in BLAST search, despite the fact that their phylogenetic relatives produce sesamin. This result is consistent with previous notions regarding plant P450 phylogeny, i.e. that proteins in the CYP81 family have not been conserved in basal angiosperms and gymnosperms27. Considering that these plants are phylogenetically very distant from Lamiales, their sesamin synthase genes, which are as yet unidentified, are thought to be independent in origin from Si_CYP81Q1 and probably occurred through CEV (Fig. 1a).
It should be noted that Si_CYP81Q1 has a unique Ala (Ala308) residue that is crucial for catalysis of MDB formation in its distal I-helix, where among P450s, a conserved Thr residue (distal-Thr) is commonly located (Supplementary Fig. 3)16,28. Each of the three Cuscuta P450s, Cc_CYP81Q110, Cc_CYP81Q111, and Ca_CYP81Q111, also has an Ala residue (Ala313, Ala310, and Ala310, respectively), at the site corresponding to the distal-Thr residue (Supplementary Fig. 3). These data indicate that Cc_CYP81Q110, Cc_CYP81Q111, and Ca_CYP81Q111 satisfy the structural requirements to be considered putative MDB-forming enzymes and most likely catalyze the production of sesamin.
Functional evaluation of Cuscuta CYP81Q genes
Structural conservation of CYP81Q1-related genes within Cuscuta spp. suggests that they have common biochemical functions in planta. To evaluate biochemical properties of the Cuscuta CYP81Q genes, recombinant proteins were co-expressed with a C. campestris cytochrome P450 reductase, Cc_CPR1 (Cc043955), in a yeast system29. When fed with (+)-pinoresinol, Cc_CYP81Q110, Cc_CYP81Q111, or Ca_CYP81Q111 formed two MDBs on the two aromatic rings of the (+)-pinoresinol and produced (+)-sesamin via (+)-piperitol (Fig. 2). The stereochemistry of the (+)-sesamin produced by the three Cuscuta CYP81Q genes was identical to that generated by Si_CYP81Q1 using (+)-pinoresinol as a substrate. Moreover, they showed trace levels of MDB-forming activity for (+)-epipinoresinol and (-)-pinoresinol. The results of LC-MS analysis revealed that the product formed from (+)-epipinoresinol is likely to be a piperitol isomer, (+)-pluviatilol, which has a single MDB, and that the product from (-)-pinoresinol is (-)-sesamin, which has two MDBs. In contrast, the third CYP81 family protein found in C. campestris, Cc_CYP81AX6, did not show MDB-forming activity for any of the pinoresinol isomers tested in this study (Supplementary Fig. 4). Collectively, the data show that the three Cuscuta CYP81Q genes encode functional PSS and provide a molecular basis for the presence of sesamin in Cuscuta spp.
Open-source RNA-seq analyses showed that the two Cc_CYP81Q genes are coordinately expressed in seedlings and flower bud clusters (Fig. 3a)30, whereas Ca_CYP81Q111 was expressed in buds, ovaries and seeds but not in germinating seedlings (Fig. 3b)3. Moreover, we detected the presence of (+)-sesamin in C. campestris in both fruits containing seeds and flower bud clusters grown on Nicotiana tabacum as a non-sesamin producing parasitic host (Supplementary Fig. 1). This is consistent with the expression profiles of the two C. campestris PSS genes. By contrast, the third homolog in C. campestris, Cc_CYP81AX6, showed negligible expression in these organs. Collectively, these results show that the sesamin found in Cuscuta plants is de novo synthesized in reproductive organs and seeds, rather than produced by the host plant and transported to Cuscuta plants.
Structural comparison of CYP81Q-related genes
The high structural similarity and functional conservation between sesame and Cuscuta CYP81Q genes prompted us to estimate the time of divergence these P450 genes using the RelTime method31. The point of divergence of sesame and Cuscuta CYP81Q1 genes was calculated to be 47.8 MYA using the proportional mode (constant rate) method calibrated to the time of divergence between sesame and olive (Olea europaea), both of which are Lamiales, which was calculated to be 81 MYA (Supplementary Table 2, Supplementary Fig. 5). By contrast, the point of divergence of sesame and Cuscuta plants was estimated to be 84 MYA16. This temporal discrepancy in the estimation of divergent time among the Cuscuta CYP81Q genes as compared with lineage speciation suggests a recent origin for the PSS genes, i.e. after speciation of Cuscuta.
We next amplified the genomic regions containing CYP81Q genes from S. indicum and C. campestris using genomic DNA as a template and specific PCR primer sets. Then, we empirically confirmed the sequence of the PCR-amplified fragments by sequencing and found that both Cc_CYP81Q110 and Cc_CYP81Q111 consist of four exons separated by introns (three introns total), as predicted based on whole-genome sequence data. In contrast, Si_CYP81Q1 has two exons with a single intron15,33. The first and the third introns in the two Cc_CYP81Q genes were highly conserved (Fig. 1c). Since the overall genomic structure of these two Cc_CYP81Q genes was also conserved in another CYP81Q-related gene, Ca_CYP81Q111 found in C. australis genome3, generation of the first and the third introns of CYP81Q1 genes in Cuscuta plants might have occurred prior to speciation of C. campestris and C. australis. Importantly, the position of the second intron of the three Cuscuta CYP81Q genes was identical to that of the intron in Si_CYP81Q1. By contrast, Cc_CYP81AX6 had only a single intron and thus, showed a different genomic structure (a single intron and two exons). These common genomic signatures among CYP81Q genes observed between distantly-related plants, i.e. Cuscuta and Sesamum, implies a shared origin for these cytochrome P450 genes.
To further evaluate genomic features of CYP81Q genes, we compared nucleotide sequence similarity of introns from sesame and dodder by CLUSTALW alignment (Table 1). The intron of Si_CYP81Q1 (170 bp) and intron 2 of the three Cuscuta CYP81Q genes (222–228 bp) shared approximately 40% nucleotide sequence identity, whereas the intron of Si_CYP81Q1 and intron 3 of Cc_CYP81Q111, which is comparable in length to intron 2 (275 bp), shared only 25% identity, a level comparable to random. These results highlight structural similarity between the intron of Si_CYP81Q1 and intron 2 of the three Cuscuta CYP81Q genes. Structural comparison among intron 1 or intron 3 of Cuscuta CYP81Q genes revealed structural divergence of Cc_CYP81Q110 from the highly homologous Cc_CYP81Q111 and Ca_CYP81Q111, each of which was predicted to have a repetitive sequence in intron I (Table 1)34. Moreover, we detected DNA transposons (Stowaway and hAT) in introns of Cuscuta CYP81Q genes (Fig. 1) 35,36. Cc_Sto1 and Cc_Sto2 were located in introns 1 and 3 of Cc_CYP81Q110, respectively, whereas Cc_hAT1 was located in intron 1 of both Cc_CYP81Q111 and Ca_CYP81Q111 (Supplementary Fig. 6). Intriguingly, the characteristic footprint sequence of Stowaway was detected in introns 1 and 3 of both Cc_CYP81Q111 and Ca_CYP81Q111 at the locations corresponding to where Cc_Sto1 and Cc_Sto2 are located in Cc_CYP81Q110.
Comparison of gene synteny in CYP81Q-related gene regions
We next compared gene synteny in the regions surrounding the CYP81Q genes in S. indicum, C. campestris, and C. australis (Fig. 4). In the S. indicum genome, Si_CYP81Q1 (SIN_1025734) is located on chromosome 9, between ATPase (SIN_1025733) and an uncharacterized protein (SIN_1025735) next to the FLOWERING LOCUS C expresser (FLX)-like gene (SIN_1025736). By contrast, Cc_CYP81Q110 and Cc_CYP81Q111 were both located in reverse orientation as compared to their neighboring genes, in the vicinity of an E3-class ubiquitin ligase (E3UbL). Specifically, Cc_CYP81Q110 is between DUF4228 (Cc046290) and a hypothetical protein (Cc046293), and Cc_CYP81Q111 is between DUF4228 (Cc015411: gene number 1, Fig. 4) and another hypothetical protein (Cc015416: gene number 9, Fig. 4, Supplementary Table 1). Moreover, these genomic features were also observed nearby the RAL50776 gene in the C. australis genome. The observation that the genomic features in introns and the region adjacent to Cc_CYP81Q111 were distinct from those of Cc_CYP81Q110 and Ca_CYP81Q111 (Figs. 1 and 4) suggests that the twin homeologs in C. campestris were brought by hybridization (allopolyploidization) between two Cuscuta plants, both of which had the ability to produce sesamin, rather than occurring through WGD of an ancient C. campestris genome (autopolyploidization).
Although multiple E3UbL genes were observed between DUF4228 (LOC109177929: gene number 1, Fig. 4) and a hypothetical protein (LOC109177844: gene number 9, Fig. 4) in chromosome 5 of the I. nil genome, there were no cytochrome P450 genes in these regions of the three Ipomoea genomes corresponding to the Cuscuta PSS genes. This is notable since the arrangement of genes between DUF4228 and LOC109177844 in the I. nil genome is structurally analogous to those between Cc046290 and Cc015411, and between Cc046293 and Cc015416, in the C. campestris genome.
Collectively, comparative genomics approaches based on gene synteny across species highlight the unique presence of CYP81Q genes in Cuscuta plants, supporting the idea that CYP81Q genes appeared in the Cuscuta genome by HGT. We cannot exclude the alternative possibility that an ancestor of I. nil had a CYP81Q1 ortholog but lost it during speciation, although aside from Cuscuta, no other Convolvulaceae plants are known to accumulate sesamin14,37.
Parasitism-mediated HGT
The observance of PSS genes with conserved genomic features and molecular functions in Sesamum and the distantly-related parasitic Cuscuta plants suggests that HGT of PSS occurred through parasitism between their ancestors. To help explore the feasibility of this model of HGT, we examined whether C. campestris is able to parasitize S. indicum by co-cultivating C. campestris with S. indicum (cv. Masekin) under laboratory conditions. The results showed that C. campestris successfully formed haustoria on the stem of S. indicum 2–3 weeks after germination of the parasites (Fig. 5a). Cross sections at the haustorium revealed that the vascular tissues of S. indicum were directly connected with haustorial vascular elements of the parasite (Fig. 5b). At 8–9 weeks after germination, C. campestris formed a flower and developed an ovary that contained fertile seeds, thereby completing its lifecycle as an obligate parasite (Fig. 5c, d). The ability to parasitize the modern sesame cultivar supports the model that parasitism by a Cuscuta of a Sesamum, or a phylogenetically related plant with PSS, facilitated HGT.
The presence of intron 2 in all of the Cuscuta PSS genes (Fig. 1c) indicates that the putative HGT event likely occurred via transfer of a genomic fragment containing an ancestral CYP81Q gene with the intron, rather than via retroposition of an intron-less mRNA. To test the possibility of transfer of a genomic fragment, we analyzed sequence similarity between the genomic regions flanking CYP81Q genes of Cuscuta plants and S. indicum, and found that they shared extremely low similarity (Supplementary Fig. 7). This result does not further support the idea of transfer of a long genomic fragment to Cuscuta and is different from HGT events reported in Orobanchaceae, in which transferred genomic fragments are typically tens of kbp in length38. Nevertheless, we cannot exclude the possibility that in Cuscuta, a long genomic fragment was inserted first and then the sequence of regulatory regions changed, probably due to lower genetic constraints in those regions than in coding regions.
Acquisition of CYP81Q genes in the genome of the ancestor of Cuscuta might also have been facilitated by RNA-mediated HGT, as has been predicted in a root parasitic plant, Striga hermonthica39. In support of this idea, we note that (1) RNA molecules harboring introns (known as intron retention; IR) have been recognized to exert biological functions in plants and mammals40,41, and (2) RNA-based HGT likely occurs more readily when mRNA abundance increases since mRNA abundance might be a determinant of long-distance mobility42, whereas cellular genomic DNA content is relatively static. To test the hypothesis that RNA-based HGT accounts for the presence of CYP81Q, we surveyed IR-RNA of CYP81Q1 using S. indicum RNA-seq data29,43,44 and found this form of the RNA among minor transcripts (< 3% of total transcripts) in seeds of cv. Masekin (Supplementary Fig. 8). We also found evidence of the three components of a plant polyadenylation signal, including a near upstream element (NUE; AAUAA) and a 1 nt variant of the NUE (AAUACA); a 1 nt variant of the far upstream element (FUE; TTGTAA); and UGUA-containing hexamers following a short poly(A) stretch45, in the 3’ sequences downstream of the stop codons of Cuscuta CYP81Q genes46 (Supplementary Fig. 9). Moreover, both Cuscuta CYP81Q111 transcripts were found to harbor a short poly(A) stretch. These sequences seemed to be structurally incomplete but might be fading genomic signatures of an ancient RNA-mediated HGT event.
Next, we used qPCR and RNA-seq to look at gene expression of Si_CYP81Q1 in the host stem, where Cuscuta specifically forms haustoria. Si_CYP81Q1 was expressed at minute levels in the non-parasitized host stem. Surprisingly, Si_CYP81Q1 was significantly induced by parasitization of C. campestris, but not by mechanical wounding (Fig. 5e-g), suggesting that induction of Si_CYP81Q1 expression in S. indicum is not a response to wounding caused by the penetration of haustoria, but a specific host defense response against the parasite. Both spliced and IR-RNA forms of Si_CYP81Q1 transcripts were induced, and the ratio of IR-RNA (intron/exon) did not change after parasitism (Fig. 5e-g). We could occasionally detect both spliced and IR-RNA forms of Si_CYP81Q1 in the stem of C. campestris that had established parasitic connections to S. indicum (Fig. 5h). Sequences of the shorter and longer amplified fragments were confirmed to be the spliced and IR forms of host Si_CYP81Q1 transcripts, respectively, indicating translocation of induced RNAs of Si_CYP81Q1 to parasite stem from the parasitic interface of the host stem through the haustorium. Long-distance movement of various types of RNA between plants has been reported in interspecific grafts47 and parasite-host plant complexes5, 48, suggesting the involvement of vascular transport of RNAs. The possibility of transport of RNAs by nonenveloped RNA viruses, which encapsidate host RNAs, has also been suggested49. Our results support an evolutionary scenario in which functional HGT of PSS into the parasite genome was mediated by parasitism-evoked locally abundant RNAs. This idea is consistent with the fact that only one Cuscuta CYP81Q gene encodes a functional lignan catalytic unit in the conserved genomic synteny among Convolvulaceae plants (Fig. 4).
Presence of both PSS and Sol-B in seeds
The presence of sesamin and its derivative lignans in the seeds of distantly related plants suggests that there are common but unknown roles for sesamin and derivative lignans in these seeds. One such role might be to prevent lipid peroxidation by exerting their (pro)antioxidative activities33, thereby preserving storage resources in oil bodies for biogenesis in the next generation. This trait conferred by seed lignans seems also to be beneficial for survival of Cuscuta seedlings, which entirely rely on the nutritive reserves stored in the endosperm until they start sucking nutrition from host plants via haustoria50. Furthermore, a sterol-binding dehydrogenase found in oil bodies, known as steroleosin (Sol), is widespread in seed plants including gymnosperms, and is involved in germination and development by regulating phytohormone sensitivity51,52. A recent report using nano-beads with affinity to sesamin identified Sol-B as a sesamin binding protein and indicated that the interaction between Sol-B and sesamin is physiologically relevant in developing seedlings53. Using S. indicum Sol-B in a BLAST search to query Cuscuta genomes, we found Sol-B-like genes (Cc036318.t1, Cc036490.t2, and C002N0117E0.1) in Cuscuta and an partial Sol-B gene amplified by PCR from genomic DNA of C. japonica that show striking structural similarity to those of Ipomoea but not those of Sesamum, roughly in line with the evolutionary plant lineages (Supplementary Fig. 10a). Thus, Sol-B genes seems to have gradually diverged along with plant speciation, again highlighting the unusual similarity among CYP81Q genes in Sesamum and Cuscuta (Fig. 1b). We further confirmed co-expression of Cc_Sol-B and Cc_CYP81Q in C. campestris seeds such as S. indicum (Supplementary Fig. 10b and c)53, suggesting common roles via interaction between sesamin and Sol-B protein in their seeds. Whether HGT-mediated metabolic traits acquired from host plants has contributed to the unique physiology and ecology of Cuscuta specifically remains an open question that can be addressed in subsequent studies.
Recent reports have described several genomic signatures of nuclear HGT from hosts to parasites but the functions of genes transferred to parasitic plants have remained largely elusive7,23,38,39,54,55. In this work, we identified that PSS genes in C. campestris and C. australis are biochemically functional and we provide a possible explanation based on genomic analysis as to how PSS genes were integrated into the genomes of Cuscuta spp. Our findings support the idea that a common ancestor of the Cuscuta and Grammica subgenera in the Cuscuta genus gained a functional genomic PSS gene from an ancestor of S. indicum through parasitism-mediated HGT, rather than, as generally assumed, by CEV, and thereby acquired (+)-sesamin biosynthetic ability (Fig. 6, Supplementary Fig. 11). Furthermore, we suggested a possible mode of parasitism-mediated HGT from the host plant to the parasitic plant via locally induced gene expression in the parasitic interface by uncovering genomic signatures of parasite Cuscuta CYP81Q genes, as well as parasitism-mediated induction and transfer of host Si_CYP81Q1 transcripts into the parasite.
Our findings herein underscore that in addition to CEV, parasitism-mediated HGT provides another possible driving force for the sporadic occurrence of specialized metabolites in the plant kingdom, as has been observed for microorganisms. Furthermore, we note that Cuscuta plants are obligate parasites and can parasitize a wide variety of host plants (Supplementary Fig. 12)6,23,56, and suggest that there might be more opportunities for HGT to occur in such parasitic plant species than in non-parasitic plants. The biological significance of the presence of (+)-sesamin-derived lignans in Cuscuta and the molecular mechanism of HGT from host to parasite remain unclear. However, evidence of parasitism-mediated HGT of PSS in Cuscuta plants provides a new perspective on metabolic evolution in plants beyond typical phylogenetic constraints and reproductive isolation, and establishes these parasites as new tools for investigating the biological activities of specialized metabolites. The unique ecological nature of Cuscuta plants as mediators of genetic and chemical information, given their promiscuous host range and ability to disperse seeds at long distance, including across oceans1,2, suggest that they might play important though as yet to be elucidated roles in plant evolution and adaptation47.
Plant materials
Cuscuta campestris seeds were germinated and seedlings were parasitized onto host plants as described previously57. Nicotiana tabacum plants were grown and parasitized by C. campestris as described previously51. The C. campestris-N. tabacum parasitic complexes were grown at 25°C under a 16/8 h light/dark cycle, and flowers and fruits of the C. campestris were harvested. The harvested tissues were frozen in liquid nitrogen and stored at -80°C. Seeds of Sesamum indicum cv. ‘Masekin’ were germinated and grown in soil (Sukoyakabaido, Yanmar, Osaka, Japan) mixed with the same volume of vermiculite (GS30L, Nittai Co., Ltd., Aichi, Japan) under natural sunlight illumination from May to August in 2019. Mature stems of 8- to 9-week-old S. indicum were parasitized by C. campestris.
Chemicals
(+)-Sesamin (Chromadex), (+)-sesaminol (Nagara Science), (+)-pinoresinol (Sigma-Aldrich), and (+)-piperitol, synthesized previously16, were prepared as the reference samples.
LC-MS analysis
Lignans in extracts of flowers and seeds of Cuscuta plants (C. campestris and C. japonica) were analyzed as follows. A 10 mg sample of lyophilized flowers or seeds was homogenized to a fine powder using a TissueLyser II (Qiagen). Next, 1 ml of 70% acetonitrile aqueous solution was added to the homogenized samples and the samples were extracted using an ultrasonic cleaner at room temperature for 2 min. The filtered extracts were analyzed using an ion-trap time-of-flight mass spectrometer (LCMS-IT-TOF, Shimadzu) equipped with a photodiode array detector (Shimadzu). Each component was separated using a YMC Triart C18 column (TA12S03-1503WT, 150 mm × 3 mm I.D., 3 µm P.S.) with mobile phases A, 0.1% HCO2H-H2O; and B, 0.1% HCO2H-MeOH, in a linear gradient elution with 30-50-90-90-30-30% B (0-10-25-32-32.01-40 min) at a flow rate of 0.3 ml/min.
PCR
Genomic DNA from C. campestris (Cc), C. japonica (Cj) and S. indicum (Si) was prepared from stems using the DNeasy Plant Mini kit (Qiagen) according to the manufacturer’s protocol. Total RNA was prepared using the RNeasy Plant Mini kit (Qiagen) from the following tissue following lysis using a TissueLyser II (Qiagen): Cc seeds, Cc seedlings (at 7 days after germination), Si stem-Cc haustorium junction (parasitic interface tissues), Cc stems, Si stems with and without mechanical wounding with a blade, and Si leaves. The RNA samples were treated with DNase Set (Qiagen) to eliminate contaminating genomic DNA. After DNase I treatment, cDNA was synthesized using an oligo dT primer and the PrimeScript RT reagent kit (TAKARA BIO). Reactions were performed according to the manufactures’ instructions. PCR products were amplified using specific primer sets (listed in Supplemental Data 4) as described previously17,58,59,60. Briefly, genomic PCR, qPCR and RT-PCR was conducted using PrimeSTAR Max DNA Polymerase (TAKARA BIO), GoTaq qPCR Master Mix (Promega) and PrimeSTAR GXL DNA Polymerase (TAKARA BIO), respectively.
RNA extraction for RT-PCR and RNA-seq
Total RNA for RT-PCR was prepared from S. indicum (Si) CYP81Q1 in C. campestris (Cc) stems and total RNA for RNA-seq was prepared from Cc seedlings, Si-Cc haustorium junctions (parasitic interface tissues), Si-Cc Si leaves, Cc stems growing on the Si stem, Si stems with and without mechanical wounding with a blade, and Si leaves. Cc seedlings were harvested 7 days after germination, which was grown under 16h light/ 8h dark cycle for 5 days, blue-light illumination for 1h, under dark condition for 23h, and a 16/8 h light/dark cycle 1 day. Si plants were used 4-6 weeks and 12-16 cm height as the parasitized plants and as the plant samples with and without mechanical wounding. After parasitization or mechanical wounding, plant samples were under blue-light illumination for 1h, under dark conditions for 23h, and a 16/8 h light/dark cycle for 5 days. The samples were harvested 6 days later after parasitization or mechanical wounding. Parasitic interface tissues were harvested from 1.5 cm length include the parasitic interface. Si stems with mechanical wounding were harvested from 1.5 cm length including the cutting point. Si stems without mechanical wounding were harvested from a 1.5 cm length of epicotyl. To collect Si leaves, the topmost portions of leaves that had grown to at least 3 cm were chosen. Cc stems were harvested at 10.5 -13.5 cm length from 1.5 cm near the parasitic interface to the Cc stem tip to avoid cross-contamination with the host. Harvested tissues were washed twice with 70% EtOH for 2 min and rinsed with nuclease-free water for 2 min to clean the surface of the tissues. Total RNA was extracted using the RNeasy Plant Mini kit (Qiagen) after lysis with a TissueLyser II (Qiagen) and then treated using the TURBO DNA-free Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. Each RNA sample was derived from a single organism.
Genome and transcriptome
We used open source Cuscuta transcriptome and genome data sets. The RNA-seq data for C. campestris were obtained from the DNA Data Bank of Japan Sequenced Read Archive accession number DRA009453 (https://trace.ddbj.nig.ac.jp/dra/index_e.html/DRA009453)30. The assembled genome sequence and annotations for C. campestris were obtained from the plaBi database (http://plabipd.de/portal/cuscuta-campestris) and for C. australis from the National Center for Biotechnology Information (NCBI) Sequence Read Archive (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA394036). Cc_CPR1, Cc_CYP81Q110, Cc_CYP81Q111, Cc_CYP81AX6, and Ca_CYP81Q111 correspond to Cc043955, Cc046292, Cc015414, Cc047366, and RAL50776 (responsible for C65N0022E0.1), respectively. The DNA-seq data for C. americana was obtained from SRA experiment ERR3569498 of BioProject PRJEB34450 and for C. californica, from SRA experiment ERR3569499. Synteny analysis was performed using BLASTP to search for best hit protein sequences in the databases indicated in Supplementary Table 3. All translation products of the genes listed in Supplementary Table 3 were used as queries to search all of the databases.
Molecular phylogenetic analysis
The nucleotide sequences of Cuscuta CYP81-related genes used in this study are listed in Supplementary Table 4 and Supplementary Data 1. The phylogenetic trees shown in Fig. 1, Supplementary Fig. 2, and Supplementary Fig. 9 were constructed using a maximum likelihood (ML) method in the Seaview software (phyML ln(L)=-23653.0, 1868 sites, GTR 4 rate classes)61. The timetree shown in Supplementary Fig. 5 was inferred by applying the RelTime method31, 62 to a phylogenetic tree whose branch lengths were calculated using the ML method and the Tamura-Nei substitution model. Evolutionary analyses were conducted in MEGA X software63.
Genome comparisons
The 10 kb genomic sequence containing the CYP81Q-related gene in the S. indicum genome was compared to CYP81Q-related genes in Cuscuta genomes using nucmer in the Mummer -v3.1.0 package64. Structural similarities were visualized with dotplots using the DNAnexus Dot browser (available at https://dnanexus.github.io/dot/) and Unipro UGENE software (http://ugene.unipro.ru/) (Supplementary Fig. 6).
Molecular cloning
Cc_CYP81Q110 (Cc15414), Cc_CYP81Q111 (Cc046292), and Cc_CYP81AX6 (Cc047366) were amplified by reverse transcription-polymerase chain reaction (RT-PCR) from cDNA prepared from a mixture of flower buds and ovary tissue from C. campestris with the specific primers described in Supplementary Table 4. Ca_CYP81Q111 was artificially synthetized without codon-optimization (Eurofins Genomics), based on the sequence of RAL50776. All of the P450 genes were cloned into yeast expression vectors and heterologously co-expressed in yeast with a C. campestris cytochrome P450 reductase, Cc_CPR1 (Cc043955), as described previously29.
Biochemical analysis
Biochemical analysis of Cuscuta Cc_CYP81Q110, Cc_CYP81Q111, Cc_CYP81AX6, and Ca_CYP81Q111 was performed basically as described previously29. Briefly, yeast cells expressing Cuscuta CYP genes were pre-cultured overnight at 30°C with rotary shaking at 120 rpm in 3 ml synthetic defined liquid medium containing a set of amino acids appropriate for the designated expression vectors. Next, 50 µl of stationary phase culture was transferred to 1 ml of fresh medium in 24-well plates supplemented with 100 µM of lignans as substrates. The cultures were further incubated for 24 h at 30°C with rotary shaking at 120 rpm. For extraction, the cells were harvested with the medium and disrupted by sonication. The homogenate (50 µl) was mixed with 50 µl acetonitrile and centrifuged at 21,000×g for 10 min. The supernatant was collected, filtered through a Millex-LH syringe filter (Merck Millipore) and subjected to the following HPLC analysis. Briefly, the filtered assay products were separated using a Cortecs UPLC C18+ column (part# 186007401, 2.7 µm, 3 mm × 75 mm, Waters) with mobile phases (A; 0.1% trifluoroacetic acid-H2O, and B; 0.1% trifluoroacetic acid-acetonitrile) in a linear gradient with 30-80-80-30-30% B (0-1.4-1.8-2.0-2.5 min) at a flow rate of 1.25 ml/min, and lignans were detected using a photodiode array detector at 280 nm.
Preparation and imaging of agarose-embedded sections
Parasitic interface tissues were fixed with 4% (w/v) paraformaldehyde in phosphate buffer solution (Fujifilm Wako Pure Chemical Corporation, Osaka, Japan), embedded in 8% (w/v) agarose, and cut using a MicroSlicerTM ZERO 1N (DOSAKA, Kyoto, Japan) into 200-μm sections. Histochemical staining of sections was performed using a 0.5% (w/v) solution of Toluidine Blue O (1B-481, Waldeck GmbH & Co., Munster, Germany) in distilled water. Stained slices were observed using a BX53 Biological Microscope (Olympus, Tokyo, Japan).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgements:
We thank D. Nelson (Univ. of Tennessee), T. Umezawa (Kyoto Univ.), Y. Tobimatsu (Kyoto Univ.), S. Yoshida (NAIST), K. Shirasu (RIKEN), H. Satake (SUNBOR), K. Shimamoto (SUNBOR), N. Okitsu (SIC), M. Takagi (SST), Y. Kawai (NCGM), M. Yamamoto (Toyama Univ.), Y. Ogata (Osaka Pref. Univ.), K. Nishitani (Kanagawa Univ.), and all the participants in the Society of Post Youth Agronomists (SPYA 2019, at Ogoto, Shiga pref. Japan) and the Frontier Research Society of Plant Specialized Metabolism (FPSM 2019, at Matsumoto, Nagano pref. Japan) for helpful comments on and support for this work. Computations were performed in part on the NIG supercomputer at ROIS National Institute of Genetics.
Funding
This work was partly supported by Grants-in-Aid for Scientific Research (18H03950 and 19H00944, JSPS to K.A.) and a Grant-in-Aid for JSPS Fellows (19J14848, JSPS to KS).
- García, M. A., Costea, M., Kuzmina, M. & Stefanović, S. Phylogeny, character evolution, and biogeography of Cuscuta (dodders; Convolvulaceae) inferred from coding plastid and nuclear sequences. Am. J. Bot. 101, 670–690 (2014).
- Costea, M., García, M., Baute, K. & Stefanović, S. Entangled evolutionary history of Cuscuta pentagona clade: A story involving hybridization and Darwin in the Galapagos. Taxon 64, 1225–1242 (2015).
- Sun, G. et al. Large-scale gene losses underlie the genome evolution of parasitic plant Cuscuta australis. Nat. Commun. 9, 2683 (2018).
- Heide-Jørgensen, H.S. Establishment of the parasite. In Parasitic Flowering Plants. pp. 263–305. Koninklijke Brill NV, Leiden (2008).
- Kim, G., LeBlanc, M. L., Wafula, E. K., dePamphilis, C. W. & Westwood, J. H. Genomic-scale exchange of mRNA between a parasitic plant and its hosts. Science 345, 808–811 (2014).
- Yang, Z. et al. Convergent horizontal gene transfer and cross-talk of mobile nucleic acids in parasitic plants. Nat. Plants 5, 991–1001 (2019).
- Zhang, D. et al. Root parasitic plant Orobanche aegyptiaca and shoot parasitic plant Cuscuta australis obtained Brassicaceae-specific strictosidine synthase-like genes by horizontal gene transfer. BMC Plant Biol. 14, 19 (2014).
- Sun, T., Xu, Y., Zhang, D., Zhuang, H., Wu, J., Sun, G. An acyltransferase gene that putatively functions in anthocyanin modification was horizontally transferred from Fabaceae into the genus Cuscuta. Plant Divers. 38:149–155 (2016).
- Donnapee, S., Li, J., Yang, X., Ge, A., Donkor, P. O., Gao, X., Chang, Y. Cuscuta chinensis Lam.: A systematic review on ethnopharmacology, phytochemistry and pharmacology of an important traditional herbal medicine. J. Ethnopharmacol. 157, 292–308 (2014).
- Ahmad, A., Tandon, S., Xuan, T.D., Nooreen, Z. A Review on Phytoconstituents and Biological activities of Cuscuta species. Biomed Pharmacother. 92, 772–795 (2017).
- Du, K.Z., Li, Z., Guo, X., Li, Y., Chang, Y.X. Quantitative Analysis of Phenolic Acids and Flavonoids in Cuscuta chinensis Lam. by Synchronous Ultrasonic-Assisted Extraction with Response Surface Methodology. J. Anal. Methods Chem. 2018, 6796720 (2018).
- Huang, R., O'Donnell, A.J., Barboline, J.J., Barkman, T.J. Convergent evolution of caffeine in plants by co-option of exapted ancestral enzymes. Proc. Natl. Acad. Sci. U.S.A. 113, 10613–10618 (2016).
- Mao, L. et al. Genomic evidence for convergent evolution of gene clusters for momilactone biosynthesis in land plants. Proc. Natl. Acad. Sci. USA 117, 12472–12480 (2020).
- Umezawa, T. Phylogenetic distribution of lignan producing plants. Wood Res. 90, 27–110 (2003).
- Ono, E. et al. Formation of two methylenedioxy bridges by a Sesamum CYP81Q protein yielding a furofuran lignan, (+)-sesamin. Proc. Natl. Acad. Sci. USA 103, 10116–10121 (2006).
- Noguchi, A. et al. Mode-of-action and evolution of methylenedioxy bridge forming P450s in plant specialized metabolism. Plant Biotechnol. 31, 493–503 (2014).
- Ono, E. et al. Formation of a methylenedioxy bridge in (+)-epipinoresinol by CYP81Q3 corroborates with diastereomeric specialization in sesame lignans. Plant Cell Physiol. 59, 2278–2287 (2018).
- Srivastava, S., Gupta, M. M., Prajapati, V., Tripathi, A. K. & Kumar, S. Sesamin a potent antifeedant principle from Piper mullesua. Phytother. Res. 15, 70–72 (2001).
- Jayasinghe, L. et al. Antifungal constituents of the stem bark of Bridelia retusa. Phytochemistry 62, 637–641 (2003).
- He, X. H. et al. Two new lignan glycosides from the seeds of Cuscuta chinensis. J. Asian Nat. Prod. Res. 12, 934–939 (2010).
- Rho, T. & Yoon, K. D. Application of off-line two-dimensional high-performance countercurrent chromatography on the chloroform-soluble extract of Cuscuta auralis seeds. J. Sep. Sci. 41, 2169–2177 (2018).
- Abu-Lafi, S. et al. Sesamin from Cuscuta palaestina natural plant extracts: Directions for new prospective applications. PLoS One 13, e0195707 (2018).
- Vogel, A. et al. Footprints of parasitism in the genome of the parasitic flowering plant Cuscuta campestris. Nat. Commun. 9, 2515 (2018).
- Akashi, T., Aoki, T. & Ayabe, S. CYP81E1, a cytochrome P450 cDNA of licorice (Glycyrrhiza echinata L.), encodes isoflavone 2'-hydroxylase. Biochem. Biophys. Res. Commun. 251, 67–70 (1998).
- Liu, C.-J., Huhman, D., Sumner, L. W. & Dixon, R. A. Regiospecific hydroxylation of isoflavones by cytochrome P450 81E enzymes from Medicago truncatula. Plant J. 36, 471–484 (2003).
- Hoshino, A. et al. Genome sequence and analysis of the Japanese morning glory Ipomoea nil. Nat. Commun. 7, 13295 (2016).
- Nelson, D.R., Schuler, M.A., Paquette, S.M., Werck-Reichhart, D. & Bak, S. Comparative genomics of rice and Arabidopsis. Analysis of 727 cytochrome P450 genes and pseudogenes from a monocot and a dicot. Plant Physiol. 135, 756–772 (2004).
- Imai, M. et al. Uncoupling of the cytochrome P-450cam monooxygenase reaction by a single mutation, threonine-252 to alanine or valine: possible role of the hydroxy amino acid in oxygen activation. Proc. Natl. Acad. Sci. USA 86, 7823–7827 (1989).
- Murata, J. et al. Oxidative rearrangement of (+)-sesamin by CYP92B14 co-generates twin dietary lignans in sesame. Nat. Commun. 8, 2155 (2017).
- Kaga, Y. et al. Interspecific signaling between the parasitic plant and the host plants regulate xylem vessel cell differentiation in haustoria of Cuscuta campestris. Front. Plant Sci. 11, 193 (2020).
- Tamura, K. et al. Estimating divergence times in large molecular phylogenies. Proc. Natl. Acad. Sci. USA 109, 19333–19338 (2012).
- Kumar, S., Stecher, G., Suleski, M. & Hedges, S. B. TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819 (2017).
- Wan, Y., Li, H., Fu, G., Chen, X., Chen, F. & Xie, M. The relationship of antioxidant components and antioxidant activity of sesame seed oil. J. Sci. Food Agric. 95, 2571–2578 (2015).
- Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
- Habu, Y., Hisatomi, Y. & Iida, Molecular characterization of the mutable flaked allele for flower variegation in the common morning glory. Plant J. 16, 371–376 (1998).
- Hoshino, A., Yoneya, Y. & Kuboyama, T. A Stowaway transposon disrupts the InWDR1 gene controlling flower and seed coloration in a medicinal cultivar of the Japanese morning glory. Genes Genet. Syst. 91, 37–40 (2016).
- Afendi, F. M. et al. KNApSAcK family databases: integrated metabolite-plant species databases for multifaceted plant research. Plant Cell Physiol. 53, e1 (2012).
- Kado, T., Innan, H. Horizontal Gene Transfer in Five Parasite Plant Species in Orobanchaceae. Genome Biol. Evol. 10, 3196–3210 (2018).
- Yoshida, S., Maruyama, S., Nozaki, H. & Shirasu, K. Horizontal gene transfer by the parasitic plant Striga hermonthica. Science 328, 1128 (2010).
- Ge, Y., Porse, B. T. The functional consequences of intron retention: alternative splicing coupled to NMD as a regulator of gene expression. Bioessays 36, 236–243 (2014).
- Yang, Y., Li, Y., Sancar, A., & Oztas, O. The circadian clock shapes the Arabidopsis transcriptome by regulating alternative splicing and alternative polyadenylation. J. Biol. Chem. 295, 7608–7619 (2020).
- Calderwood, A., Kopriva, S. & Morris, R. J. Transcript abundance explains mRNA mobility data in Arabidopsis thaliana. Plant Cell 28, 610–615 (2016).
- Wang, L. et al. Genome sequencing of the high oil crop sesame provides insight into oil biosynthesis. Genome Biol. 15, R39 (2014).
- Wang, L., Yu, J., Li, D. & Zhang, X. Sinbase: an integrated database to study genomics, genetics and comparative genomics in Sesamum indicum. Plant Cell Physiol. 56, e2 (2015).
- Shi, Y. & Manley, J.L. The end of the message: multiple protein-RNA interactions define the mRNA polyadenylation site. Genes Dev. 29, 889–897 (2015).
- Loke, J.C., Stahlberg, E.A., Strenski, D.G., Haas, B.J., Wood, P.C. & Li, Q.Q. Compilation of mRNA polyadenylation signals in Arabidopsis revealed a new signal element and potential secondary structures. Plant Physiol. 138,1457–1468 (2005).
- Xoconostle-Cázares, B., Xiang, Y., Ruiz-Medrano, R., Wang, H.L., Monzer, J., Yoo, B.C., McFarland, K.C., Franceschi, V.R., & Lucas, W.J. Plant paralog to viral movement protein that potentiates transport of mRNA into the phloem. Science 283, 94–98 (1999).
- Bera, S., Yamaguchi, K., Shigenobu, S., & Aoki, K. Trans-species small RNAs move long distances in a parasitic plant complex. Plant Biotechnol. (Tokyo) 38, 187–196, (2021).
- Routh, A., Domitrovic, T. & Johnson, J.E. Host RNAs, including transposons, are encapsidated by a eukaryotic single-stranded RNA virus. Proc. Natl. Acad. Sci. U.S.A 109, 1907–1912 (2012).
- Olszewski, M., Dilliott, M., García-Ruiz, I., Bendarvandi, B. & Costea, M. Cuscuta seeds: Diversity and evolution, value for systematics/identification and exploration of allometric relationships. PLoS One 15, e0234627 (2020).
- Lin, L.J., Tai, S.S., Peng, C.C., & Tzen, J.T. Steroleosin, a sterol-binding dehydrogenase in seed oil bodies. Plant Physiol. 128, 1200–1211 (2002).
- Li, F., Asami, T., Wu, X., Tsang, E.W. & Cutler, A.J. A putative hydroxysteroid dehydrogenase involved in regulating plant growth and development. Plant Physiol., 145, 87–97 (2007).
- Tera, M. et al. Identification of a binding protein for sesamin and characterization of Its roles in plant growth. Sci. Rep. 14, 8631 (2019).
- Yang, Z. et al. Horizontal gene transfer is more frequent with increased heterotrophy and contributes to parasite adaptation. Proc. Natl. Acad. Sci. USA 113, E7010–E7019 (2016).
- Zhang, C. et al. Horizontal gene transfer has impacted cox1 gene evolution in Cassytha filiformis. J. Mol. Evol. 88, 361–371 (2020).
- Hettenhausen, C. et al. Stem parasitic plant Cuscuta australis (dodder) transfers herbivory-induced signals among plants. Proc. Natl. Acad. Sci. USA 114, E6703–E6709 (2017).
- Hozumi, A. et al. Arabinogalactan proteins accumulate in the cell walls of searching hyphae of the stem parasitic plants, Cuscuta campestris and Cuscuta japonica. Plant Cell Physiol. 58, 1868–1877 (2017).
- Narukawa, H., Yokoyama, R., Kuroha, T. & Nishitani, K. Host-produced ethylene is required for marked cell expansion and endoreduplication in dodder search hyphae. Plant Physiol. 185, 491–502 (2021).
- Ono, E., et al. Glycoside-specific glycosyltransferases catalyze regioselective sequential glucosylations for a sesame lignan, sesaminol triglucoside. Plant J. 101, 1221–1233 (2020).
- Shimizu, K., Hozumi, A. & Aoki, K. Organization of vascular cells in the haustorium of the parasitic flowering plant Cuscuta japonica. Plant Cell Physiol. 59, 720–728 (2018).
- Gouy, M., Guindon, S. & Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224 (2010).
- Tamura, K., Tao, Q. & Kumar, S. Theoretical foundation of the RelTime method for estimating divergence times from variable evolutionary rates. Mol. Biol. Evol. 35, 1770–1782 (2018).
- Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
- Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004).
Table 1: Structural comparison of CYP81Q-related genes
- OnoetalSupplFig1.jpg
- OnoetalSupplFig1continued.jpg
Supplementary Fig. 1: Lignans in Cuscuta plants Chemical structure of lignans identified from Cuscuta plants in this study are shown. Red, methylenedioxy bridges (MDBs) on aromatic rings. MS chromatograms at m/z=381.1240, 379.1090, and 377.0938 showing values of sodium adduct ions corresponding to pinoresinol (1), piperitol (2), and sesamin (3) in fruits (top) containing seeds, and in flower bud clusters (middle) from Cuscuta campestris along with a standard (bottom). Retention times of (+)-pinoresinol, (+)-piperitol, and (+)-sesamin standard samples (bottom) were 17.38, 22.58, and 26.75 min, respectively. In contrast, we did not detect pinoresinol (1), piperitol (2), or sesamin (3) in seeds of C. japonica. MS chromatograms at m/z=687.1790 and 849.2310 showing values of sodium adduct ions corresponding to two lignan diglycosides (cuscutoside A (4) and cuscutoside B (5)) and a lignan triglycoside, cuscutoside D (6). MS chromatograms corresponding to these cuscutosides were detected in fruits (top) containing seeds, but not in flower bud clusters (bottom). The metabolic profile is consistent with the presence of sugar-modified lignans; however, we were unable to confirm these chemical structures due to a lack of available standards for authentication. Various lignans, including pinoresinol and sesamin, were previously reported to be found in C. chinensis3. Sesamin was also reported in C. australis and C. palaestina4,5,6.
- OnoetalSupplFig2.jpg
Supplementary Fig. 2: Phylogenetics of Cuscuta CYP81Q-related genes NGS data (SRA IDs ERR3569498, ERR3569499, SRR2143215, ERR3651373, and ERR3569501) for five Cuscuta spp. (C. americana, C. californica, C. gronovii, C. europaea, and C. epithymum) were assembled using Trinity ver.2.9.17. Contigs showing the highest similarity with Cc_CYP81Q110/Cc_CYP81Q111 were selected by BLASTn search for each Trinity assembly. With this approach, we failed to obtain sequences with structural similarity to Cuscuta CYP81Q-related genes from de novo assembled contigs derived from C. reflexa (ERR3528105, SRR1171084, and SRR2142348), C. japonica (ERR3528106, ERR3580100, ERR3580101, and the contigs previously published by Ikeue et al. 20158) or C. monogyna (ERR3569504) of the subgenus Monogynella (connected by a dotted line). Due to incomplete reconstruction of Cuscuta CYP81Q genes, a consensus sequences corresponding to the exon 2 were used for the following phylogenetic analysis (the exon 2 nucleotide sequence for CYP81Q-related genes are available in Supplementary Data 2). Phylogenetic clade (A-to-S) is labeled with species names as defined in a previous phylogenetic study9. a. A phylogenetic tree was constructed by maximum likelihood using Seaview software (phyML: ln(L)=-1333.8, 387 sites, bootstrap=100 rep., GTR 4 rate classes)10. The numbers at the branches indicate bootstrap values. The sequence of Si_CYP81Q1 exon 1, which corresponds to exon 2 of Cuscuta CYP81Q-related genes, was used as the outgroup. The three exon 2 sequences from subgenus Grammica (C. americana, C. californica, and C. gronovii) were co-clustered with those of C. australis and C. campestris, while the two sequences from subgenus Cuscuta (C. europaea and C. epithymum) formed a distinct phylogenetic clade from that of Grammica, roughly in line with the phylogeny of Cuscuta spp9. These results support the ideas that 1) CYP81Q-related genes are widely conserved among the subgenera Grammica and Cuscuta, 2) the origin of this gene likely ascends to a monophyletic ancestor located between subgenera Monogynella and Cuscuta, and 3) the genes underwent nucleotide diversification from the common ancestral CYP81Q orthologue along with speciation of subgenera Cuscuta and Grammica. Dynamic chromosomal changes such as hybridization and WGD that likely occurred multiple times during speciation of Cuscuta spp.9 make its phylogeny complicated and controversial. For example, there are discrepancies in the phylogenies between the plastid and nuclear genomes. Therefore, while various data suggest that C. campestris is a hybrid species, there is no clear evidence so far regarding the parental species11. Since it seems that the phylogeny of the PSS gene matches fairly well with that of Cuscuta plants, the evolutional analysis of this HGT-derived gene might provide an opportunity to trace Cuscuta phylogenies that recently underwent complicated speciation events. Although Cc_CYP81Q110 and Cc_CYP81Q111 are most likely twin homeologs derived from the predicted WGD in C. campestris11, Cc_CYP81Q110 is more distant from Cc_CYP81Q111 as compared to Ca_CYP81Q111. This phylogenetic topology of PSS genes suggests two possible evolutionary views: 1) that Cc_CYP81Q110 underwent either neo-functionalization that is yet to be characterized or non-functionalization by accumulating polymorphic sites due to a release from functional constraints of the ancestral PSS activity after WGD, or 2) that C. campestris is a hybrid species with two ancestral genomes, one that is closely related to C. australis and harbors an ortholog gene of Cc_CYP81Q111, and the other an unidentified species in subgenus Grammica that shares an ortholog of Cc_CYP81Q110. Further reciprocal characterization of CYP81Q-related genes and comparative genomics among Cuscuta spp. should help fully unravel the evolutionary path taken by C. campestris. b. Genomic PCR for exon 2 of Cuscuta CYP81Q genes. A specific band was amplified from gDNA from C. campestris (Cc, subgenus Grammica) but not C. japonica (Cj, subgenus Monogynella) (top), whereas an internal control gene, RPS-18, was amplified from both (middle). Genomic DNAs used for PCR templates (bottom).
- OnoetalSupplFig3.jpg
Supplementary Fig. 3: Multiple alignment of CYP81Q-related proteins A multiple alignment of CYP81Q-related proteins was executed using the muscle method with Seaview software10. Arrow, position of distal-Thr residue in the I-helix of cytochrome P450 monooxygenase, which is required for the full monooxygenase reaction12. PSS proteins of Sesamum and a phylogenetically related species (Phryma leptostachya) commonly possess Ala residue at this position, rather than a Thr residue (e.g. Ala308 in Si_CYP81Q1)13. Notably, the Si_CYP81Q1-A308T mutant form, in which the Ala308 residue is replaced with Thr, displays significantly decreased levels of MDB-forming catalysis, demonstrating that this Ala residue is crucial for MDB formation by PSS. Similarly, Cuscuta CYP81Q genes also have an Ala residue at this position (Ala310 in Cc_CYP81Q110; Ala313 in Cc_CYP81Q111 and Ala310 in Ca_CYP81Q111), whereas two other S. indicum P450s, Si_CYP98A20 (coumarate 3-hydroxylase) and Si_CYP92B14 (sesamolin and sesaminol synthase), which are also involved in the phenylpropanoid pathway, have a distal-Thr residue14. Thus, the Ala residue commonly observed in Cuscuta and Sesamum CYP81Q genes suggests a genetic signature for a functional PSS enzyme.
- OnoetalSupplFig4.jpg
Supplementary Fig. 4: Enzymatic activity of Cc_CYP81AX6 (Top) Negative control (empty yeast expression vector, pYE22m) for the bioconversion assay. Only the tested substrate (blue) was observed. (Middle) No product was observed in assays in which Cc_CYP81AX6 was co-expressed with Cc_CPR1. (Bottom) In reference assays in which Si_CYP81Q1 was co-expressed with Si_CPR1, (+)-pinoresinol was converted to (+)-sesamin (red), as shown in a previous report14. These results not only indicate that Cc_CYP81AX6 has no PSS activity but also provide a possible explanation as to why I. nil does not accumulate (+)-sesamin even though it has genes that are structurally similar to Cc_CYP81AX6 in its genome. a, (+)-pinoresinol; b, (+)-sesamin; c, (-)-pinoresinol; d, (-)-sesamin; e, (+)-epipinoresinol; f, (-)-epipinoresinol.
- OnoetalSupplFig5.jpg
Supplementary Fig. 5: Timetree of CYP81Q-related genes A timetree inferred by applying the RelTime method9, 23 to a phylogenetic tree whose branch lengths were calculated using the maximum likelihood (ML) method and the Tamura-Nei substitution model. The timetree was computed using the following calibration constraints, 81 MYA is set as the split calibration time between Olea europaea and Sesamum indicum, and under a normal distribution (σ0.25) based on the estimated divergence time in timetree (Supplementary Table 2)16. The estimated log likelihood value of the tree is -19275.40. This analysis was done using 18 nucleotide sequences. Codon positions included were 1st+2nd+3rd+noncoding. There were a total of 1735 positions in the final dataset. Evolutionary analyses were conducted using MEGA X software17. Numbers indicate estimated time of divergence (MYA).
- OnoetalSupplFig6.jpg
Supplementary Fig. 6: Transposons detected in the intron of CYP81Q-related genes a. Insertion sites of Stowaway transposons. TA dinucleotide target site duplication is indicated in boldface. Putative footprints of the Stowaway elements are underlined. Numerals indicate positions in the intron sequences. b. Comparison of the conserved TIR sequences of Stowaway transposons from Ipomoea nil18 and Cuscuta plants. c. Comparison of entire sequences of Cc_Sto1 and Cc_Sto2. Terminal inverted repeat sequences are underlined. d. Insertion sites of hAT transposons. Putative 8-bp target site duplication is indicated in boldface. Numerals indicate positions in the intron sequences. Due to the low similarity of the target sequence region between CYP810Q110 and CYP810Q111, it is possible that the hAT element inserted into Cc_CYP81Q111 and later exited19. If this is the case, the underlined sequence could be a footprint of a hAT element. e. Comparison of TIR sequences of hAT transposons from Ipomoea purpurea18 and Cuscuta plants. f. Comparison of entire sequences of hAT elements from Cuscuta plants. Terminal inverted repeat sequences are underlined.
- OnoetalSupplFig7.jpg
Supplementary Fig. 7: Genome-by-genome alignment of genomic regions containing CYP81Q-related genes Structural similarity in a broad genomic region (~10 kb with CYP81Q in the center) was evaluated using two different dot-plotting tools. (left) Pair-wise dot plotting indicates genomic regions with > 70% nucleotide identity in each 100 bp window between two genomes, as performed using Unipro UGENE20. Structural similarity between Cuscuta and Sesamum CYP81Q-related genes was observed only in exon regions, whereas overall similarity was observed among Cuscuta sequences. (right) Alignment of Cuscuta genomic sequences versus Sesamum indicum genomic sequence, performed using nucmer in the Mummer -v3.1.0 package21. The plots show structural similarity of S. indicum with C. campestris and C. australis when the S. indicum genome was used as a reference (x-axis) (upper) and when the C. australis genome was used as a reference (x-axis) (lower). Blue dots, unique forward alignments; orange dots, repetitive alignments. Arrows indicate the approximate locations of the CDSs in the CYP81Q gene of each reference. Sca, scaffold. Structural similarities were visualized with dot plots using the DNAnexus Dot browser (available at https://dnanexus.github.io/dot/), reproducing the same results found with UGENE (left), i.e. that only exons share structural similarity. These results are consistent with the idea that either 1) non-coding regions outside of PSS genes change easily due to loosened constraints for nucleic acid alteration as compared to the coding region, or 2) a small genomic fragment including the PSS ORF was transferred in a parasitism-mediated HGT event.
- OnoetalSupplFig8.jpg
Supplementary Fig. 8: Investigation of intron retention (IR) transcripts of Si_CYP81Q1 in sesame seeds a. RNA-seq data from S. indicum seeds of four cultivars (SRR1055252, SR1055260, SRR5429392, and SRR5429398) were mapped on the S. indicum genome using the HISAT2 aligner22. Identification of a small portion of RNA reads covering the intron of the CYP81Q1 gene (NP_001306620), as indicated by the dotted red box, supports the idea that IR-transcripts of CYP81Q1 gene are expressed as minor transcripts. b. Reads covering exon-intron junctions and reads in the intron are minor transcripts, accounting for < 3% of CYP81Q1 transcripts. We counted numbers of reads (1) covering exon 1-exon 2, (2) mapping to exons, (3) covering exon 1-intron, (4) covering intron-exon 2, and (5) mapping to the intron, and then calculated splicing efficiency (6) using the following formula: (2)/{(2)+[(3)+(4)]}/2. Intron retention (7) was estimated as 1-(6). Relative abundance of spliced transcripts (6), black; of IR-transcripts (7), gray (graph on the right). These results suggest the presence of a very small portion of IR-CYP81Q1 transcripts. Seed developmental stage was described in Ono et al., 200623. DAG, days after gemination treatment; DPA, days post anthesis.
- OnoetalSupplFig9.jpg
Supplementary Fig. 9: Genomic DNA alignment of 3’-untranslated regions (UTR) of Cuscuta CYP81Q genes to mRNA (Left) Genomic sequences of 3’-UTR of the three Cuscuta CYP81Q genes and corresponding transcripts were aligned using the muscle algorithm in Seaview software10. Within c.a. 350 bp downstream of the stop codon, there are putative poly(A) signal-like sequences (orange boxes): (A) near upstream element (NUE: AAUAAA), (B) 1nt-variant (AAUACA) of NUE, (C) 1nt-variant (TTGTAA) of far upstream elements (FUE), (D) UGUA hexamers24, and (E) poly(A)-like short stretch24. Asterisks indicate the putative transcription end points (PTEPs). The PTEPs for Cc_CYP81Q110 and Cc_CYP81Q111 were estimated using C. campestris RNAs (ERR1916350-ERR1916364) mapped on the C. campestris genome (PRJEB19879) using HISAT2 aligner22; reliable mapping regions of Cc_CYP81Q110 and Cc_CYP81Q111 were then extracted as putative mRNAs by Stringtie after disabling the transcript trimming option25. Similarly, the PTEP of Ca_CYP81Q111 (C65N0022E0.1) was estimated using C. australis RNAs (SRR6664648-SRR6664654) from the C. australis genome project (PRJNA394036). Notably, the poly(A)-like short stretches (E) observed in the three genomic regions of Cuscuta CYP81Q genes are included in their putative transcripts. (Right) Graphical images of mRNA read mapping on the genome sequence of Ca_CYP81Q111 (top), Cc_CYP81Q110 (middle), and Cc_CYP81Q111 (bottom). Dotted rectangles, 3’-UTRs of each Cuscuta CYP81Q gene.
- OnoetalSupplFig10.jpg
Supplementary Fig. 10: Characterization of steroleosin B-related (Sol-B) genes a. Phylogenetic tree of steroleosin-B (Sol-B)-related proteins Sol-B (also annotated as 11-beta-hydroxysteroid dehydrogenase-like 5) was identified to be a sesamin binding protein that accumulates in seeds26. A phylogenetic tree was constructed using the maximum likelihood method in Seaview software (phyML: ln(L)=-10012.9, 1026 sites, LG 4 rate classes)10. Sesamum Sol-like sequences were isolated from RNA-seq datasets of S. indicum (PRJNA350858) and S. radiatum (PRJNA633647) by executing a BLAST search with query sequences from S. indicum Sol-A (AAL13315) or Sol-B (AAM46847)14,27. Because only the partial CDS sequence was obtained from the primary assembled contigs for the S. radiatum Sol-B-like gene, we virtually constructed a putative full-length sequence by in silico elongation of the partial sequence using the VP-seq method28. Similarly, C. japonica Sol-B-like sequence was obtained from RNA-seq datasets of C. japonica (DRX027870-027874). The amino acids sequences from S. radiatum and C. japonica are provided in Supplementary Data 3. The scale bar indicates 0.2 amino acid substitutions per site. Numbers at branch points indicate bootstrap values. Cuscuta Sol-B-like genes clustered with those of phylogenetically-related relatives, Ipomoea and Solanum plants in the Solanales order and apart from those of Sesamum spp. in the Lamiales order, in contrast with to the phylogeny of CYP81Q genes as shown in Fig. 1. As the topology of the tree roughly follows plant evolutionary lineages at the order level, these genes are likely to have diverged along with plant speciation rather than arising or changing via HGT. Sol-B-related genes from Cuscuta, yellow; from Sesamum, green. An outer clade including Sesamum Sol-A and Arabidopsis thaliana HSD1 formed an outgroup next to the Sol-B-related clade via a steroleosin of the gymnosperm Pinus massoniana (pine). Thus, the tree topology supports the idea that Sol-A and Sol-B diverged prior to speciation of angiosperms. Notably, C. campestris has two Sol-B-like genes, one of which (Cc036490t2) is structurally similar to that of C. australis. This result is analogous to the phylogenies of Cuscuta CYP81Q genes (Fig. 1, Supplementary Fig. 2), likely mirroring the characteristic genomic architectures of C. campestris. Amino acid sequences of steroleosin-related proteins used for phylogenetic analysis are available in Supplementary Data 3. b. Cuscuta Sol-B gene was amplified from genomic DNA from C. japonica (Cj) and C. campestris (Cc). c. qPCR analysis revealed the presence of transcripts of both Cc_Sol-B and Cc_CYP81Q genes in developing seeds of C. campestris. Expression levels were normalized to Cc_TUB (Cc032064) 29.
- OnoetalSupplFig11.jpg
Supplementary Fig. 11: Three possible models for the sporadic distribution of specialized metabolites (left) Model 1: Lineage-specific gene loss or functional differentiation (open triangle) within metabolic radiation derived from a common biosynthetic gene (black triangle) that evolved in the ancestral plant results in a patchy pattern of absence of the metabolite (black circle). As an example, functional differentiation of Sesamum PSS specifically occurred in CYP81Q3, and results in the absence of sesamin and the presence of other lignans, namely alatumin and 2-episesalatin30. (middle) Model 2: Convergent evolution (CEV) is the main cause of the observed sporadic presence of common metabolites (black circle) and occurs when distantly related plants independently acquire biosynthetic genes with similar functions (black triangle and square), as in the case for caffeine biosynthesis genes in coffee, tea, and cacao plants21 and momilactone biosynthesis genes in rice and a bryophyte, Calohypnum plumiforme31. Biosynthetic genes that appear through CEV generally share little or moderate structural similarity to one another due to their distinct genetic origins. (right) Model 3: Parasitism-mediated HGT is a third possible explanation for a sporadic distribution of metabolites (black circle) in common among different plants, which in this model would be generated by related genes (black triangle) shared among host and parasitic plants. Because they share a common genetic origin, the respective biosynthetic genes have high similarity and structurally similar genes are rarely found in close relatives of the parasite.
- OnoetalSupplFig12.jpg
Supplementary Fig. 12: Spontaneous co-parasitism of Cuscuta plant on various plants Left: photo of spontaneous co-parasitism by a Cuscuta campestris plant for two different plants, tobacco (Nicotiana tabacum) and Arabidopsis thaliana (ecotype: Col.) in our laboratory condition. Multiple adjacent hosts are spontaneously parasitized by a single Cuscuta plant. Cuscuta bridge is considered to facilitate plant-to-plant signaling such as herbivory-induced defense signals32. Right: spontaneous co-parasitism of Cuscuta plants is often observed in nature (July 2019 at Hirosaki, Aomori-Pref. Japan) and is consistent with the wide host range of Cuscuta14,33. The finding that Cuscuta plants can parasitize multiple adjacent hosts suggests that they might fill an ecological role as mediators of genetic and chemical information between heterologous plants and further, that they might play important but as yet unappreciated roles in plant evolution and adaptation.
- OnoSupplementaryData1f.txt
Nucleotide sequences of P450 genes for phylogenetic analysis (Figure 1, Supplementary Figure S5) (fasta files)
- OnoetalSupplemenatryData2f.txt
Nucleotide sequences of exon 2 of CYP81Q-related genes for phylogenetic analysis (Supplementary Figure S2) (fasta files)
- OnoetalSupplementaryData3f.fa
Supplementary Data 3: Amino acid sequences of steroleosin-related proteins for phylogenetic analysis (Supplementary Figure S10) (fasta files)
- OnoetalSupplementaryTablesf.xlsx
Supplementary Table 1: Comparison of CYP81Q-related genes to Cc_CYP81Q110
Supplementary Table 2: Estimated divergence times of Cuscuta and other plants Data matrix constructed using Timetree (http://www.timetree.org/)1. The estimated time of divergence (dark gray) between Ipomoea nil and Cuscuta australis was based on a genome analysis of Cuscuta campestris2.
Supplementary Table 3: Gene synteny with functional annotation Genomic resources (database and URL) and gene accession numbers/gene IDs with functional annotation for genes shown in Fig. 4.
Supplementary Table 4: Sequences of oligonucleotide primers used in this study
- SupplementaryReferences.docx
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}