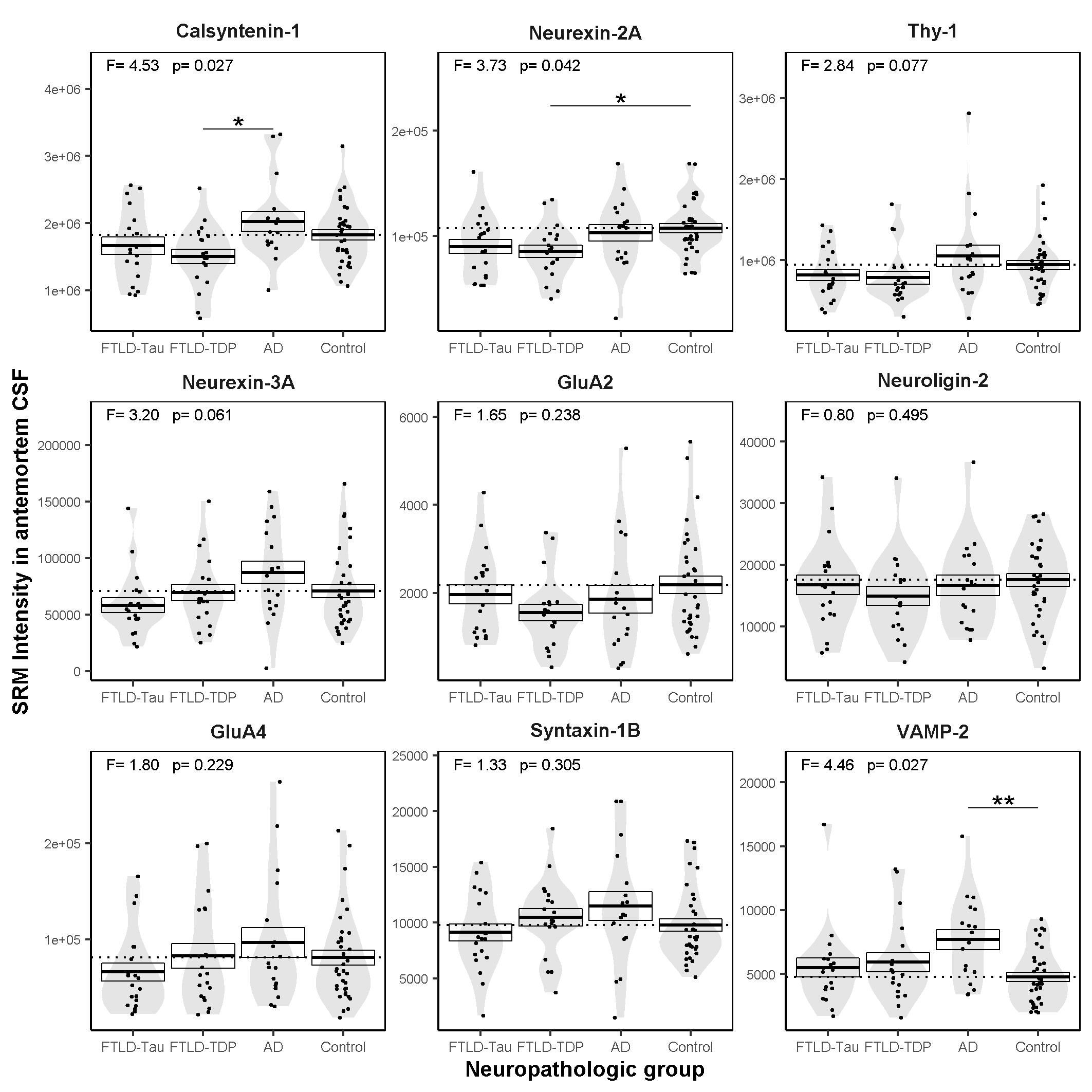
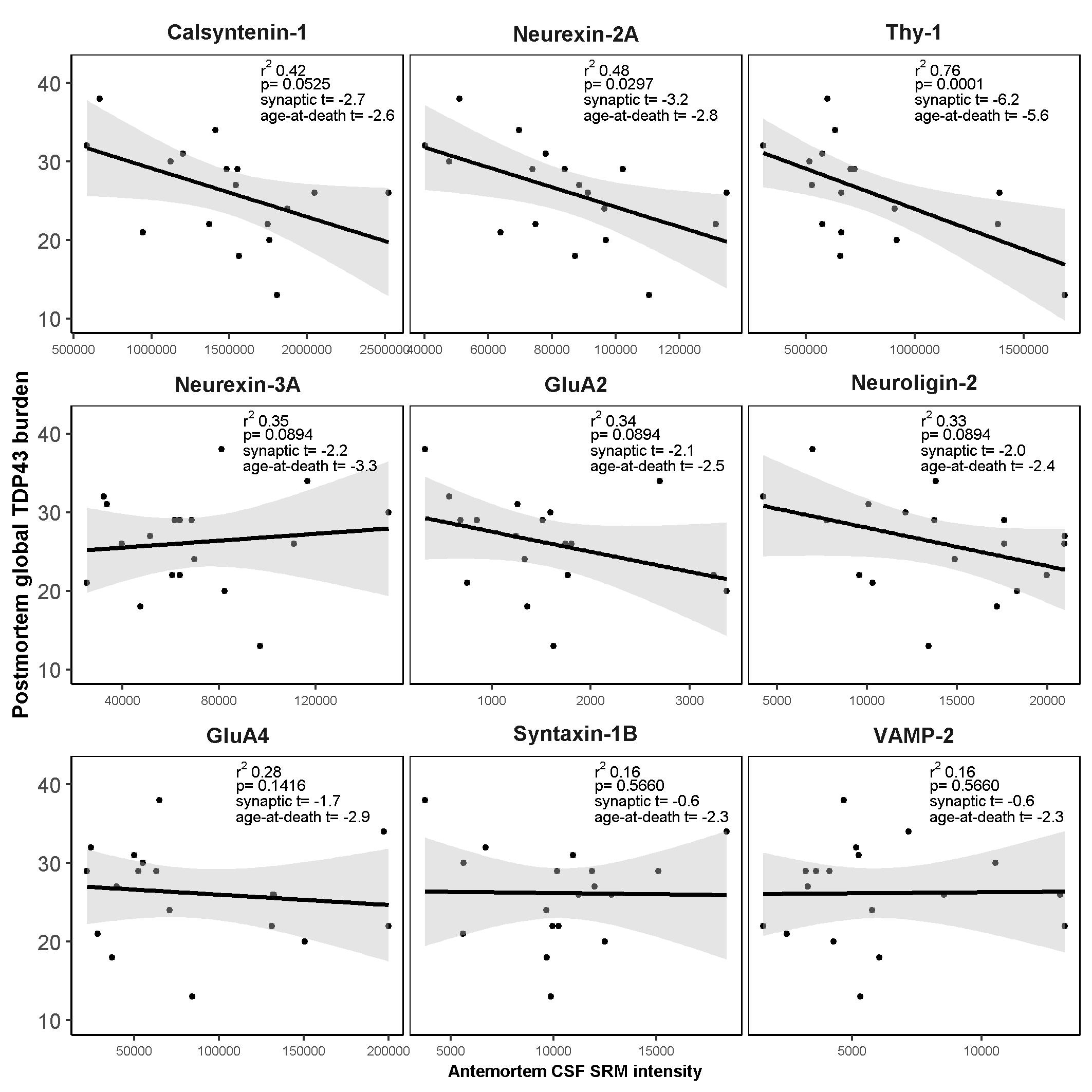
Here we report a comprehensive evaluation of the antemortem CSF levels of a panel of 9 synaptic proteins in a neuropathological cohort of FTLD (FTLD-Tau and FTLD-TDP), AD and cognitively normal controls. CSF levels of calsyntenin-1 and neurexin-2a were correlated in all patient groups and were lower in the FTLD-TDP neuropathological subtype compared to neuropathologically confirmed AD and cognitively normal controls. Furthermore, antemortem CSF levels of calsyntenin-1 and neurexin-2a inversely correlated with global TDP-43 burden post-mortem. Neither protein was altered in FTLD-Tau compared to controls or FTLD-TDP nor associated with post-mortem global tau burden. In summary, this study shows that low antemortem levels of calsyntenin-1 and neurexin-2a are related to global post-mortem TDP-43 burden in FTLD.
To our knowledge, this is the first study to relate CSF levels of neurexin-2a or calsyntenin-1 to the FTLD-TDP subtype or post-mortem TDP-43 burden. A previous study has reported an association between low CSF calsyntenin-1 levels in patients with behavioural variant FTD (bvFTD) compared to cognitively normal controls and compared to presymptomatic carriers of C9orf72, GRN or VCP mutations [40]. As the pathology underlying clinically defined FTLD syndromes is heterogeneous, the results of the previous study cannot be directly compared to the current study; clinical diagnoses of bvFTD is split between FTLD-TDP and FTLD-Tau subtypes and the small number of cases with a bvFTD diagnosis in the current study (n = 10) prohibits meaningful comparison across syndromes. Nevertheless, both studies point towards altered abundance of calsyntenin-1 in CSF from patients clinically diagnosed with FTLD syndromes and those with neuropathologically confirmed FTLD compared to cognitively normal controls.
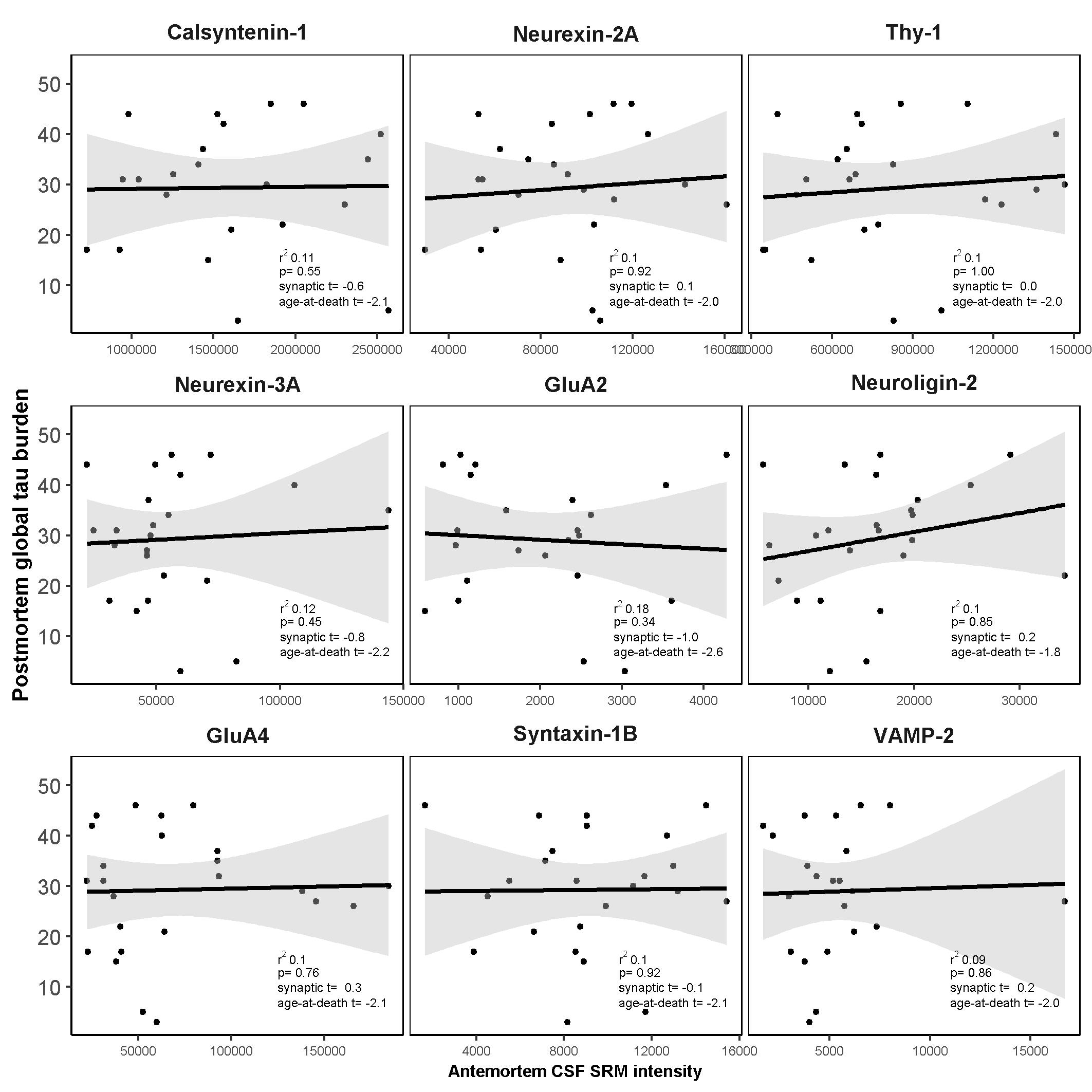
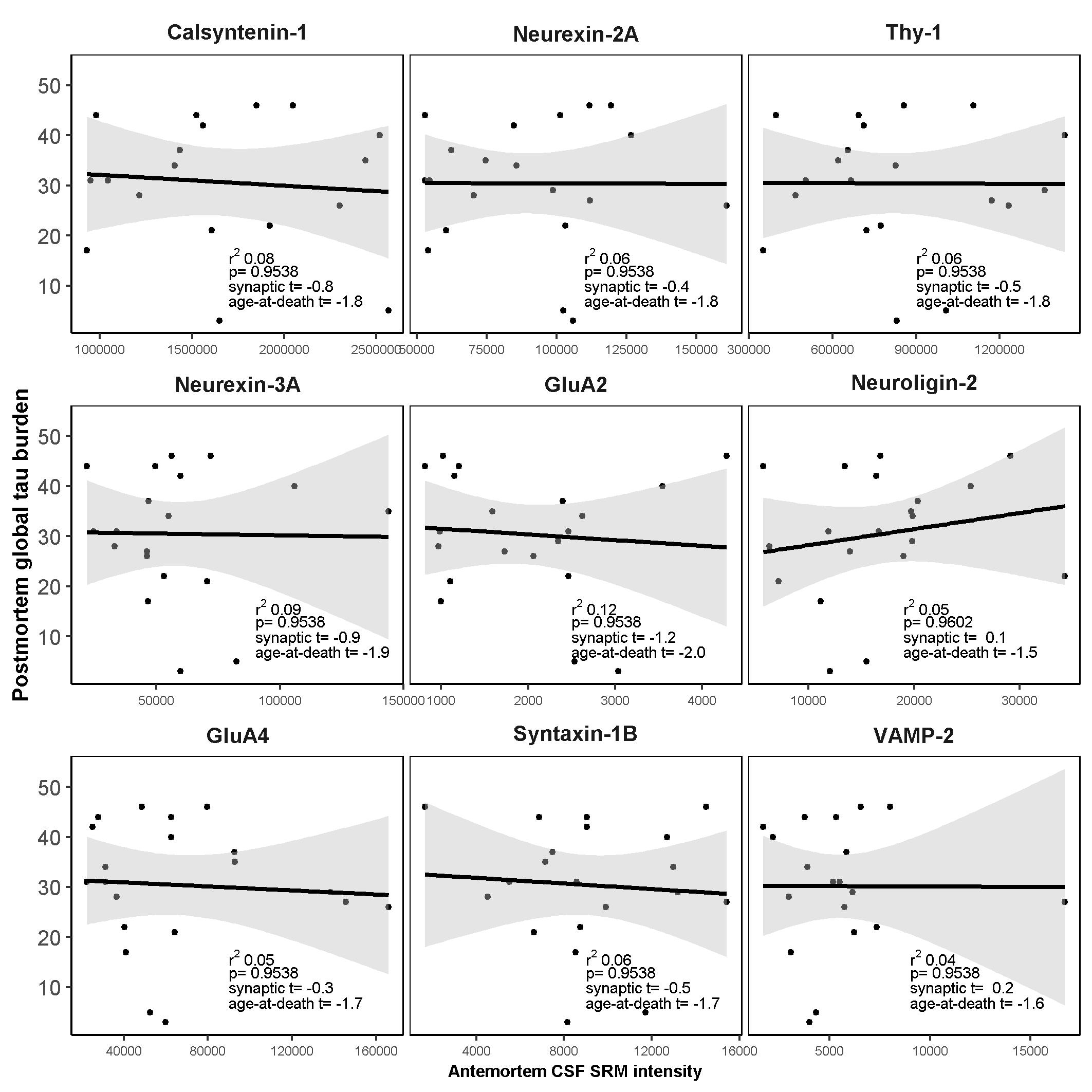
Another important finding of this study is that while AD neurofibrillary tangle pathology (Braak score < 2 [35]) was present in 17% of the FTLD-Tau and 24% of the FTLD-TDP patients and TDP-43 pathology was present in 28% of the AD patients, removal of these participants from the analyses had little impact on the findings. The degree of association (model r2) of calsyntenin-1 and neurexin-2a with postmortem global TDP-43 burden was marginally elevated in cases without AD co-pathology, which could suggest a negative confounding effect of neurofibrillary tangle pathology. However, this difference could also be attributed to the reduced sample size. Taken together with the lack of association between antemortem CSF levels of these two proteins and postmortem tau burden, we propose that changes in antemortem CSF levels of calsyntenin-1 and neurexin-2a in FTLD are related to underlying TDP-43 burden in the brain, and are largely independent of the presence of AD-related tau pathology.
A differential relationship of CSF levels of these synaptic proteins in the context of tau pathology is particularly intriguing in light of our previous studies in clinical AD cohorts. Using the same SRM method described here [13], we previously reported low CSF calsyntenin-1 and neurexin-2a in cognitively normal subjects with a biomarker profile indicative of stage 1 preclinical AD compared to cognitively normal subjects with no evidence of AD pathophysiology in CSF. We also observed relatively elevated levels of both proteins in subjects with prodromal AD (mild cognitive impairment with AD biomarker profile) and AD dementia compared to controls [13]. We have since replicated this nonlinear CSF profile over the AD continuum in a cohort of adults with Down syndrome across the AD clinical spectrum [41]. Others have also reported elevated CSF calsyntenin-1 in subjects with mild cognitive impairment and AD dementia compared to cognitively normal controls [42]. In the current study, CSF calsyntenin-1 levels were only nominally elevated in pathologically confirmed AD patients compared to controls, and neurexin-2a levels were comparable between AD and controls. Nevertheless, there is growing evidence in clinical cohorts of sporadic AD and Down syndrome that elevated CSF calsyntenin-1 and neurexin-2a occur concomitantly with elevated levels of CSF tau markers (phosphorylated and total tau). One interpretation of this finding is that reduced CSF calsyntenin-1 and neurexin-2a in preclinical AD may reflect reduced synaptic density in individuals who already show signs of brain amyloidosis, and that this effect is confounded by widespread tau-mediated neurodegeneration at later disease stages. In support of this, the current study shows that the highest levels of both neurexin-2a and calsyntenin-1 were found in the AD group, while the lowest levels were found in the FTLD-TDP group (with little or no tau pathology). Thus these data support the hypothesis that low CSF calsyntenin-1 and neurexin-2a may be confounded to a certain extent by the presence of tau pathology, particularly in AD.
Why CSF levels of neurexin-2a and calsyntenin-1 were correlated across patient groups and showed a stronger association with the FTLD-TDP subtype compared to the other synaptic markers is the subject of further investigation, but could be due to differences in function and/or regional expression patterns in the brain. Studies in mice have shown a functional interaction of α-neurexins to calsyntenin-3 that regulates excitatory synaptic innervation [43–45] but a functional interaction between calsyntenin-1 and neurexin-2a has yet to be determined. Interestingly, both proteins are subject to proteolytic processing by α- and γ- secretases [46].
Calsyntenin-1 is abundant in most neurons of the CNS [47] where it has been shown to modulate postsynaptic calcium signalling and promote dendritic spine assembly [19]. We quantified calsyntenin-1 using two proteotypic peptides (GNLAGLTLR, IISTITR) as internal standards, both of which are located in the extracellular domain common to both calsyntenin-1 isoforms. More specifically, they are located in sAlcα, a product of α-secretase (ADAM10/17) proteolytic cleavage of the mature calsyntenin-1 protein [19]. In our previous shotgun analysis of human CSF [13], we identified 63 calsyntenin-1 peptides, all of which were located in sAlcα, suggesting that sAlcα is the predominant calsyntenin-1domain present in CSF. This is consistent with other studies that report that the sAlcα peptide and the Alcα-p3 (product of γ-secretase) are released into the CSF [19, 42], while the remaining transmembrane stump is internalized into the spine apparatus. It has yet to be determined whether CSF levels of sAlcα reported here reflect brain levels of the sAlcα fragment only, the full-length calsyntenin-1 protein or both.
The neurexins are a complex family of proteins that are generated from three different genes (NRXN1, NRXN2, NRXN3) with alternative promoters (α, β, γ) and extensive alternative splicing resulting in over 1000 distinct neurexin isoforms [48]. Here we quantified neurexin-2a using three proteotypic peptides (LQGDLSFR, LGERPPALLGSQGLR, LSALTLSTVK) all of which are located in the extracellular domain that is liberated by α-secretase (ADAM10/17) cleavage. The remaining neurexin C-terminal fragment (γ-secretase substrate) accumulates mainly at the presynaptic terminals [46]. Each individual neurexin isoform is associated with specific neuronal cell types. In rodents, neurexin-2a is highly expressed in pyramidal cells and parvalbumin interneurons [48, 49]. The formation of a trans-synaptic complex with their post-synaptic partners, the neuroligins, allows the structural assembly of excitatory synapses, by triggering the recruitment of postsynaptic NMDA and AMPA glutamate receptors [17, 50]. Neurexins have also been shown to facilitate the calcium-dependent release of synaptic vesicles [51–53]. Deletion of the NRXN2α gene in mice results in reduced social and repetitive behaviors in a sex-specific manner but were not associated with cognitive deficits [54]. In humans, NRXN2 mutations have been associated with altered risk of autism spectrum disorders [55], schizophrenia [55] and early-onset epileptic encephalopathy [56].
There is growing evidence for an involvement of the glutamatergic system in FTLD syndromes [57]. In relation to FTLD-TDP, maintenance of synaptic functions in excitatory glutamatergic neurons within the brain and spinal cord may be critical for TDP-43 biological function [58] and manipulation of TDP-43 expression levels in a primary rodent neuronal culture model caused significant defects in dendritic branching and outgrowth [59]. Further studies are needed to determine whether the shared role of calsyntenin-1 and neurexin-2a in structural assembly of glutamatergic synapses can explain the low levels we observed in FTLD-TDP patient CSF and the correlation with TDP-43 burden.
From a clinical perspective, neurexin-2a gave an AUC of .75 to distinguish FTLD-TDP from controls and calsyntenin-1 showed an AUC of .76 to distinguish FTLD-TDP from AD. Despite this modest diagnostic performance, the potential clinical utility of synaptic markers likely lies in disease prognosis, monitoring of disease progression and potentially as a measure of drug response in clinical or trial settings rather than as diagnostic biomarkers. The association of antemortem CSF levels of these markers with postmortem TDP-43 burden is therefore very promising and opens the door to future studies in longitudinal clinical cohorts.
It should be noted that CSF thy-1 also correlated with calsyntenin-1 and neurexin-2a in all patient groups. We observed low CSF thy-1 in FTLD-TDP compared to AD and only a nominal decrease compared to controls. Thy-1 showed a stronger association with FTLD-TDP pathology compared to calsyntenin-1 and neurexin-2a but showed nominally the worst diagnostic performance (AUC < 69%). Thy-1 is a glycosylphosphatidylinositol-linked integral membrane protein of the immunoglobulin superfamily and component of synaptic vesicles [24]. In vitro studies indicate an important role of thy-1 in the regulated vesicular release of neurotransmitter at the synapse [24]. Taken together, thy-1 also warrants further study to better understand its clinical utility in FTLD.
Study Limitations
A limitation of this work is the relatively small study size compared to other CSF biomarker studies. However, CSF samples from well-annotated autopsy cases of these uncommon conditions are scarce and the neuropathological confirmation reduces the substantial heterogeneity inherent in clinical FTD cohorts. It should also be noted that the mean time from CSF to death was 5 years in FTLD-Tau and 4 years in the FTLD-TDP subtypes, reaching up to 10 years in some cases. This long interval and variability could influence the relationship between participants’ CSF biochemical signature and their final neuropathological findings. Finally, our control participants lack neuropathological confirmation. However, complete clinical and neuropsychological evaluations were performed to exclude significant medical (and specifically neurological) conditions in these participants.
{kind=link}
{kind=link}
{kind=link}
{kind=link}