Cell culture and materials
The human glioma cell lines U251, T98G, IDH1(R132H) U87, IDH1(WT) U87 and HEK-293T cells were purchased from ATCC and used in the present study. Cells were maintained in the medium recommended and supplemented with 10% FBS in a 37°C and 5% CO2 incubator. Recombinant lentiviral vectors were constructed with an Invitrogen ViraPower™ Lentiviral System (Carlsbad) in our laboratory.[1] The lentiviral vectors pLenti6-mCherry/NDRG2, PAX2 and PMD2G were transfected into HEK-293T cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Glioma cells were infected with viral medium from HEK-293T cells 48 h after transfection.
MTT assay
Infected cells were seeded in 96-well plates in triplicate at a starting density of 1×104 cells/well and cultured in the recommended medium without glutamine. Treated cells were washed and incubated with tetrazolium salt (MTT, 100 µg/ml; Sigma) at 37°C for 4 h. The supernatant was removed, and 150 µl of dimethyl sulfoxide (DMSO) was added to each well. The absorbance (OD) of the reaction solution at 490 nm was recorded.
Colony formation assay
Infected cells were seeded into 60-mm dishes at a density of 400 cells per dish. The cells were grown for 2 weeks in culture medium without glutamine. Then, the colonies were fixed and stained with crystal violet.
Western blotting analysis
For Western blotting analysis, total protein was prepared from human liver cell lines and clinical hepatocellular carcinoma tissue samples. Immunoblotting was performed according to standard procedures with monoclonal rabbit anti-PC antibody (Abcam ab126707, 1:2000), monoclonal mouse anti-NDRG2 antibody (Abnova H00057447-M03, 1:1000), monoclonal mouse anti-Flag antibody (Sigma F3165, 1:1000), monoclonal mouse anti-Myc tag antibody (Abcam ab32, 1:1000), monoclonal mouse anti-HA tag antibody (Abcam ab18181, 1:1000), monoclonal mouse anti-IDH1(R132H) antibody (Sigma ASB4200548, 1:200), monoclonal mouse anti-α-tubulin antibody (Boster M03989-2, 1:1000) and monoclonal rabbit anti-β-actin antibody (Boster BM3873, 1:1000).
Quantitative real-time PCR
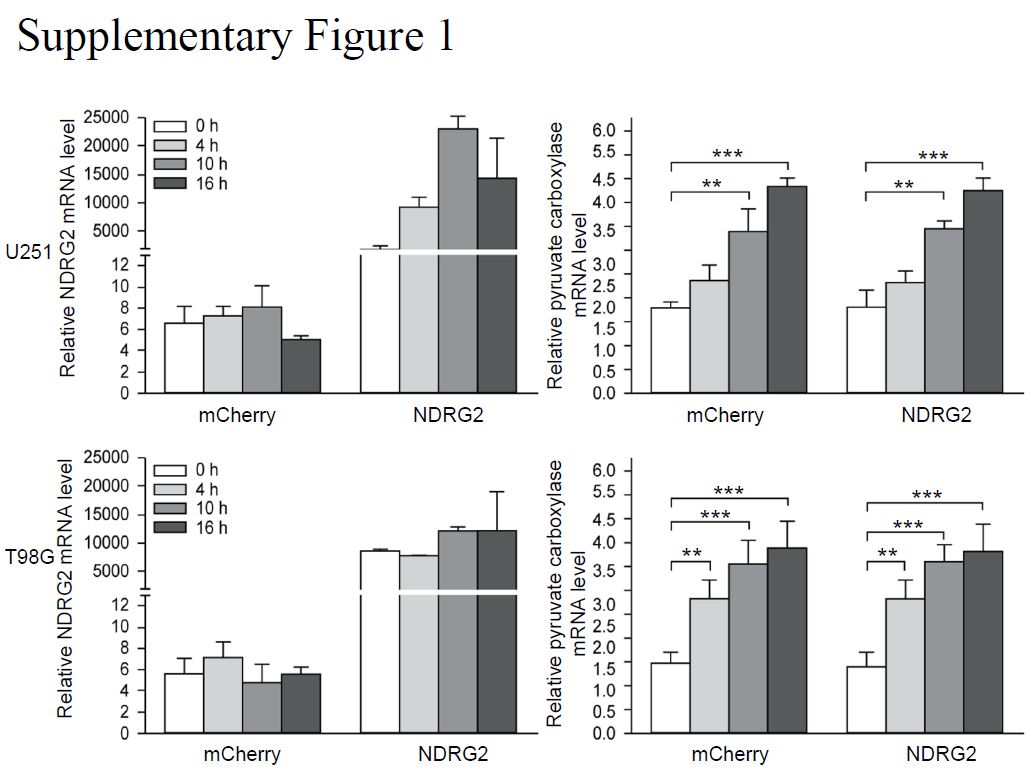
Total RNA was isolated from cells using TRIzol Reagent (Invitrogen), and then complementary DNA (cDNA) was synthesized using AMV reverse transcriptase (Promega) according to the manufacturer’s instructions. cDNA was used as a template for quantitative real-time PCR using an ABI Prism 7500 real-time PCR instrument (Applied Biosystems). The primers used for real-time quantitative PCR are listed in Table S1.
Vector construction
The construction scheme of the expression vector used for tandem affinity purification (TAP) is shown in Fig. 3A. For construction of the expression vector, the coding genes of NDRG2 were amplified by PCR and confirmed by DNA sequencing, and then target genes were ligated with a fusion expression vector containing S-tag, Flag and streptavidin-binging peptide (SBP). The primers used for PCR are listed in Table S2.
Tandem affinity purification
HEK293T cells were transfected with an SFB-tagged NDRG2 or empty vector. Twenty-four hours post-transfection, the cells were lysed in NETN buffer (20 mmol/L Tris-HCl, pH 8.0, 100 mmol/L NaCl, 1 mmol/L EDTA, 0.5% Nonidet P-40, 50 mmol/L b-glycerophosphate, 10 mmol/L NaF, and 1 mg/mL pepstatin A) at 4°C for 3 hours. The supernatant was collected for incubation with streptavidin Sepharose beads (GE Healthcare Sciences) at 4°C overnight. The next day, the beads were washed with NETN buffer five times and then eluted with 2 mmol/L biotin (Sigma) for 1 hour at 4°C twice. The elution products were incubated with S protein agarose beads (Novagen) at 4°C overnight, and after three washes, the products bound to S-protein agarose beads were subjected to SDS-PAGE and analyzed by mass spectrometry (MS).
Coimmunoprecipitation
Cells were harvested and lysed in IP buffer (50 mmol/L Tris-HCl, pH 7.4, 150 mmol/L NaCl, 1% Triton X-100, 1% sodium deoxycholate, and 1% protease inhibitor cocktails) on ice for more than 15 minutes. Cell lysates were centrifuged for 10 minutes at 13,000 rpm at 4°C, and the supernatant was transferred to a new tube. The supernatant was incubated with primary antibodies against Myc or Flag and protein A/G agarose beads (Thermo Fisher Scientific) with gentle rocking at 4°C overnight. The next day, the pellet was washed six times with IP buffer on ice and then subjected to Western blotting analysis.
Immunohistochemistry
Glioma specimens were histologically diagnosed, and glioma tissue microarrays (TMAs) were produced by the Department of Pathology, Xijing Hospital, Fourth Military Medical University. Tissue microarray (TMA) staining was performed using standard immunohistochemistry procedures. The slides were incubated overnight with primary antibodies against NDRG2 (Abnova H00057447-M03, 1:500) or PC (Abcam ab229267, 1:500). Staining intensity was scored in a blinded fashion: 1 = weak staining at ×100 magnification but little or no staining at ×40 magnification; 2 = medium staining at ×40 magnification; 3 = strong staining at ×40 magnification. The final staining index was calculated using the following formula: staining intensity × percentage.
Tumor tissues from nude mice were collected on day 28, excised and fixed with 4% formalin, and embedded in paraffin. For immunohistochemistry, 5 µm-thick tissue sections were cut, dewaxed in xylene, and rehydrated. For Ki67 staining, the slides were incubated with 1% bovine serum albumin in PBS at room temperature for 1 h for blocking and then stained with primary antibodies against NDRG2 (Abnova H00057447-M03, 1:500), PC (Abcam ab229267, 1:500) or Ki-67 (Invitrogen, PA5-19462, 1:1000) at room temperature for 4 h. The slides were subsequently washed three times with PBS to remove excess primary antibody and then incubated with anti-mouse HRP-conjugated IgG (Boster BM3895, 1:500) for 1 h at room temperature. Finally, the slides were washed three times, incubated with DAB peroxidase substrate (Sigma) and covered with glass cover slips. The staining results were observed with a bright field microscope.
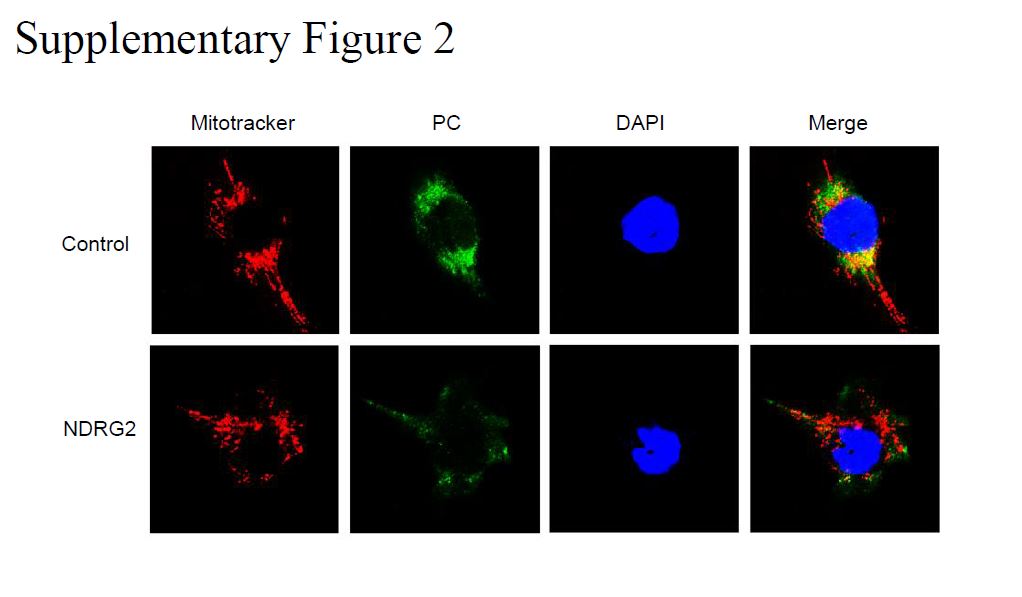
For NDRG2 and PC subcellular localization, the cells were fixed in a freshly prepared solution of 4% paraformaldehyde, rinsed, and permeabilized with 0.1% Triton X-100 in PBS. Permeabilized cells were then incubated with horse serum in PBS to block nonspecific binding. After thorough rinsing with PBS, the cells were incubated overnight with NDRG2 or PC antibody, and incubated with fluorescein isothiocyanate (FITC)-conjugated anti-rabbit antibody or Cy5-conjugated anti-mouse antibody. Dual-color detection was performed by confocal laser scanning microscopy after treatment with 4',6-diamidino-2-phenylindole (DAPI) for 10 min to label nuclear DNA.
Pyruvate carboxylase activity
Treated cells were seeded on 6-well plates at a density of 1×106 cells per well and the culture medium was changed to low glucose DMEM without phenol red (Thermo Fisher Scientific). The activity of pyruvate carboxylase in the culture medium was measured after incubation of cells for 24 h with a pyruvate carboxylase activity assay kit (Jiancheng Bioengineering). The activity of pyruvate carboxylase was normalized to the cell numbers. The cell numbers were calculated and analyzed using a Cellometer Mini bright field automated cell counter (Nexcelom Bioscience).
Tracer studies in cell cultures
U251 cells transduced with NDRG2 or mCherry were incubated in DMEM with 10 mM 13C-glucose and in the absence of glutamine for 18 hours, quenched in cold acetonitrile, and extracted in acetonitrile/water/chloroform (v/v 2:1.5:1). Metabolite fractions from cells were analyzed by mass spectrometry (MS) as previously reported [22].
In vivo tumorigenicity assay
The animal study and experimental protocols were approved by the Institutional Laboratory Animal Center at the Fourth Military Medical University. The animals were maintained and handled in accordance with the Guidelines for Accommodation and Care of Animals. JC Wang has a license for animal experiments. All mice were housed under standard conditions of a 12-hour light/dark cycle and access to food and water ad libitum. Four-week-old athymic mice were injected subcutaneously with 1×106 cells. U251 cells expressing the mCherry control were injected into the left flank, and U251 cells expressing NDRG2 were injected into the right flank. When the tumor size reached an average of 60 mm3, the mice were treated with 15 mg/kg L-DON or PBS 3 times per week. The tumor size was measured every other day, and tumor volume was calculated using a standard formula: tumor volume (mm3) = width (mm2) × length (mm) × 0.5. Eleven days after drug treatment, the mice were sacrificed and the tumors were harvested for Western blot analysis.
Statistical analysis
Statistical analysis was performed with SPSS software (version 17.0; SPSS). The results are presented as the mean ± SEM from at least three individual experiments for each group. Student’s t-test was used to compare the differences between two groups. Pearson product-moment correlation was used to calculate the correlation between NDRG2 and PC staining index in IDH1 wild-type and IDH1-R132H mutant glioma TMAs. Statistical significance was defined as p < 0.05, and statistical graphics were prepared with Origin 6.0 (Microcal Software, Inc., Northampton).
{kind=link}
{kind=link}