Drug discovery is the basis of the modern pharmaceutical market, and encompasses most of the industry’s research and development funding [1]. On average, it takes 12–15 years and $985million to deliver a drug to market, demonstrating the exhaustive time and effort required to complete the drug discovery process [2, 3]. Drug-Target Interaction (DTI) analysis is one of the most critical parts of drug discovery, and it involves calculating the binding affinity between a target protein and a ligand molecule so that appropriate ligand candidates for drugs can be chosen. These ligand candidates go on to be included in in vitro experimentation in order to identify lead compounds for the final drug. The affinity of a ligand to bind with a protein depends on the atomic interactions between the ligand and the binding region (referred to as the “binding pocket”) on the protein (Fig.1) [4].
Calculating the binding affinity between a protein and ligand can be completed through Virtual Screening (VS), where compounds are screened and binding affinity calculated using software [5] (Fig.2). The “Scoring Function”, which is the function used to calculate binding affinity, is critical for VS. Machine Learning (ML) algorithms have demonstrated considerable promise as a scoring function compared to other standard function types [6]. Given a set of training data, ML algorithms are able to learn pharmaco-like features from protein-ligand models through supervised learning functions. This allows them to accurately predict the binding affinity based on learned features that have statistically high influence [7–9, 11]. However, ML algorithms “overfit”, or learn patterns that do not correlate to a physical phenomena but still decrease error by chance [7–9, 11, 12]. This reduces their ability to generalize to out-of-distribution (OOD) data, making them unreliable for analyzing novel ligand candidates [7]. It is necessary to uncover underlying relationships between the features of protein-ligand data in order to inform the development of ML models that experience less overfitting [8].
Supervised learning techniques used to predict binding affinity can also analyze features, yet the results suffer from inconsistency and unreliability due to the overfitting of their parent algorithms [13, 14]. In comparison, unsupervised learning techniques such as Principal Component Analysis (PCA) are effective at identifying important features from protein-ligand models without overfitting because they are not designed to only minimize prediction error [15, 17]. t-Distributed Stochastic Neighbor Embedding (t-SNE) is also useful at visualizing the features of proteins due to its ability to retain high-dimensional information in low-dimensional space [16]. However, unsupervised learning has not been applied to analyze the differences between protein-ligand complexes in regards to their binding affinity. This is a research gap that can be filled to help develop ML models which learn protein-ligand feature patterns without considerably overfitting.
Objectives:
There is a pressing need to reliably identify specific biomechanical features that influence binding affinity and quantify their effect on ML performance. Current literature either suffer from drawbacks in reliability and consistency caused by supervised learning or do not specifically analyze the variance in binding affinity caused by protein/ligand features. The objectives of this study are three-fold: 1) Discover the presence of underlying biomechanical interactions that influence binding affinity, 2) Identify specific pharmaco-like features responsible for high variance in binding affinities, and 3) Determine the effect of these features on the performance of ML models in predicting binding affinity.
Gathering a greater understanding of which features influence binding affinity is necessary for designing ML models that do not overfit to training data and interpret noisy features as important patterns. Models will thereby be more generalizable to OOD data, and more successful at identifying lead compounds for inclusion in innovative drugs.
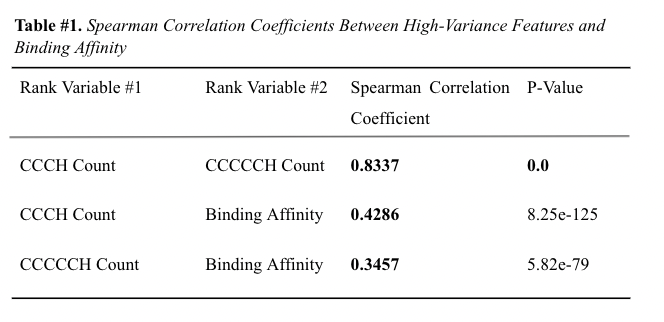{kind=link}
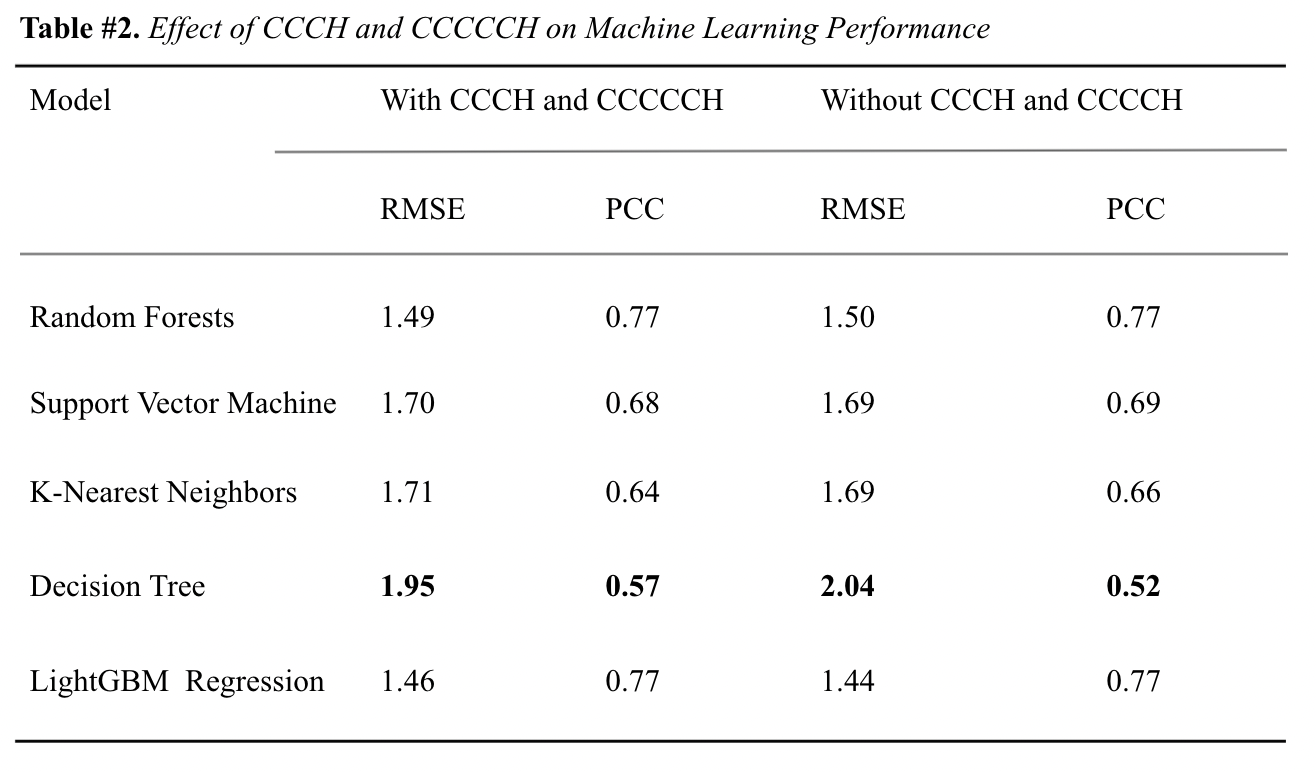{kind=link}