Considering the increasing trend in big data generation and rapid advancements in bioinformatics tools, re-analysis of pre-existed datasets for discovering new pathways and molecular keys in pathological conditions is of utmost importance. In this study, a systematic approach was adopted to generate a map of key events during the progression of HN (Fig. 1). Different proteomics datasets were entirely explored and two datasets generated by Kenneth et al. were chosen for further analysis. The datasets were proteomics profiles of tubules (PXD002106), as well as the inner and outer cortex (PXD012889) in a mouse model of HN known as 2K1C. In this model, unilateral renal artery constriction with a clip decreased renal artery perfusion and consequently systemic hypertension. Based on these profiles, the investigators have underscored the pivotal roles of Periostin, Transgelin and Vimentin in HN pathogenesis.
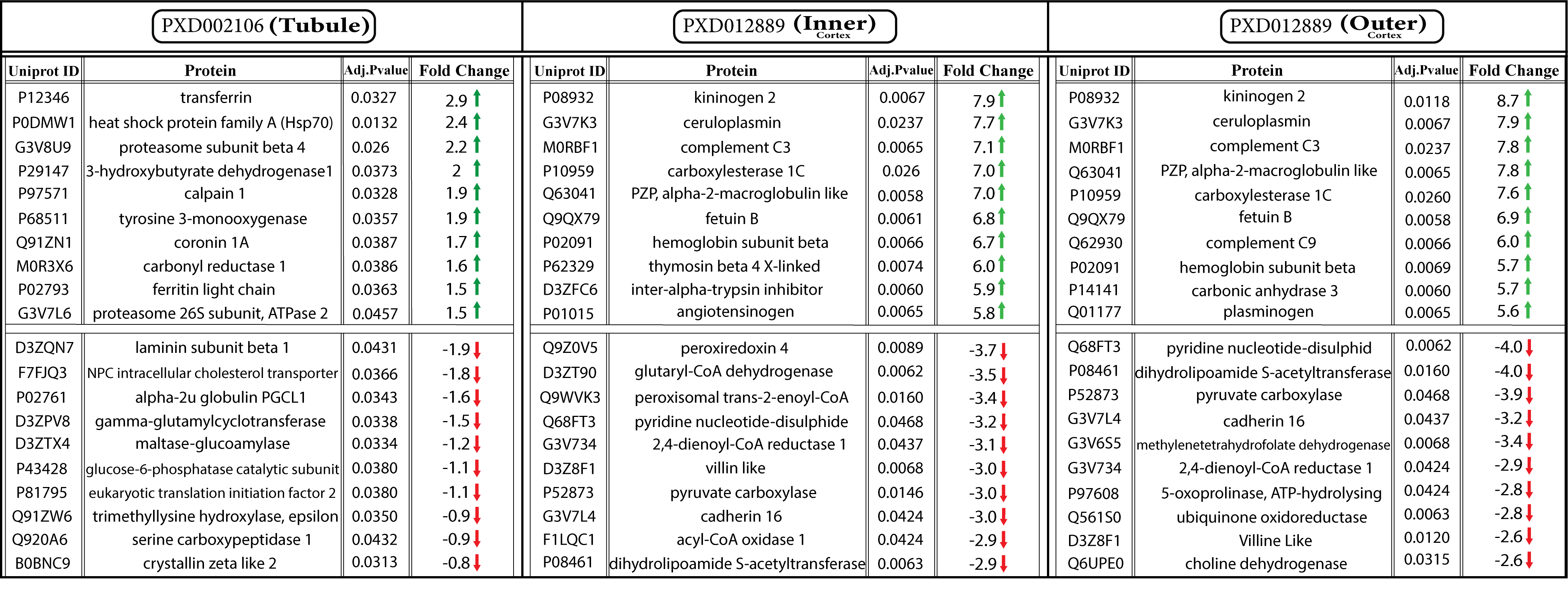
To provide further insights into the molecular pathogenesis, the above-mentioned datasets were re-analyzed with a systemic unsupervised approach. Using MaxQuant, we identified 2074, 1925, and 2015 proteins (FDR ≤ 0.01) in the tubule, inner, and outer cortex sub-compartments, respectively (Supplementary Table 1). Since we have previously highlighted the inappropriate quality of the majority of omics datasets, quality control assessment was performed before further analysis (20) Accordingly, principle component analysis (PCA) was applied which showed that most samples were scattered according to their experimental groups, indicating the acceptable quality of the data (Fig. 2a). However, a few samples not following this segregation pattern were eliminated to enhance the quality of the data. The quality of datasets was also examined by hierarchical clustering which was in agreement with the PCA (Fig. 2b). In the following, Perseus was applied to determine the differentially expressed proteins (DEPs) among the identified proteins considering permutation-based FDR ≤ 0.05. The results indicated 166, 378, and 320 DEPs in the tubule, inner and outer cortex, respectively (Fig. 2c, Supplementary Table 2). In agreement with previous studies, the alteration range of DEPs in the cortex was higher than that in the tubule (5)(6) implying the significant involvement of cortex in this disease (Table 1).
To explore the functional role of DEPs, their protein classes were determined. Notably, a considerable fraction of the DEPs was enzymes, providing an initial clue on the key role of enzymatic pathways in the disease pathogenesis (Fig. 3). Furthermore, Gene ontology (GO) enrichment analysis was carried out on different levels, including biological process (BP), molecular function (MF), and cellular component (CC) (Fig. 3). In agreement with the above findings, a variety of metabolic pathways were enriched in all sub-compartments, especially in the cortex. Furthermore, terms related to the cytoskeleton were among the other significantly enriched terms.
To further validate the above findings on the role of metabolic processes pathway enrichment analysis was conducted on the DEPs utilizing IPA. According to the GO results, a considerable fraction of the enriched pathways was related to metabolic processes especially for the inner cortex (Fig. 4). Additionally, the Rho related signaling pathways were enriched in all sub-compartments. Notably, several previous studies have reported the role of these pathways in vasoconstriction and hypertension (21–23). Moreover, other significantly enriched pathways such as VEGF signaling, complement system, and pathway of inositol have been previously recognized to be involved in kidney failure and hypertension (24–28). “HIPPO signaling” is also among the enriched pathways whose role in HN has not yet been determined.
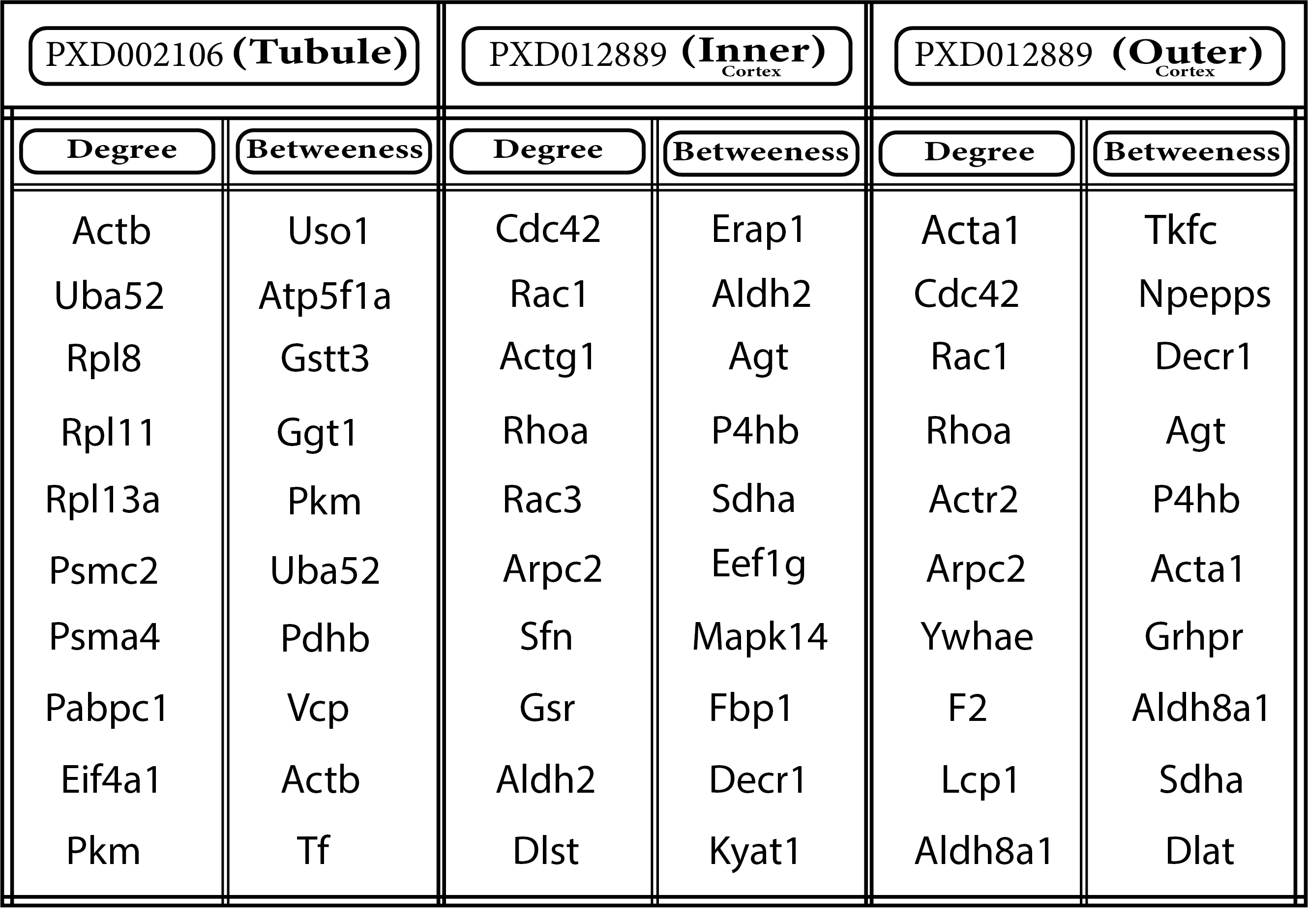
To identify the interaction map of the DEPs, PPI networks were constructed for all sub-compartments using Cytoscape CluePedia plugin. Since the central genes in networks are the key drivers of biological pathways and processes (29), the topology of the constructed networks was analyzed and central proteins were identified in terms of degree and betweenness centrality (Table 2). Noteworthy, a substantial number of the central DEPs, such as Kyat1, Decr1, Fbp1, Sdha are enzymes involved in metabolic pathways. This could further signify the critical role of metabolic processes in the progression of HTN. Moreover, two modules with the highest clustering coefficient scores were identified in each of the three networks to identify densely connected sub-graphs (Fig. 5).
In agreement with the above findings, the proteins in the modules of the inner cortex network mainly attributed metabolic processes and cytoskeleton organization. One of the outer cortex modules is also related to cytoskeleton .Notably, the other module of the outer cortex and one of the modules in the tubule network are highly rich in proteasome elements which is in line with the previous research on the renoprotective effects of proteasome inhibitors in HN (30).Remarkably, the other group of highly-connected proteins in tubule involved in energy metabolism which can be described by increased mitochondrial function in tubules in HN as also reported in previous animal models (31)(32).
To elucidate the interactions between proteins and other bio-molecules, especially metabolites, integrated networks composed of DEPs, unique enzymes, reactions, and metabolites were constructed (Fig. 6). For this purpose, among all DEPs, unique enzymes involving in specific pathways were identified and their related metabolites were added to explore the relationships between enzymatic and non-enzymatic proteins with the metabolic reactions and metabolites. Moreover, pathway and functional enrichment analyses were carried out for the predicted metabolites using MetaboAnalyst. These analyses underscored the importance of amino acid and purine metabolism as well as energy homeostasis in this disorder (Fig. 7).
As the above analyses propose the essential role of amino acid and purine metabolisms in the pathogenesis of hypertensive nephropathy, a few representatives of such processes were further investigated in more detail; Several enzymes in tryptophan degradation such as Kmo, Kyat1, Kynu, and Afmid are among downregulated DEPs. Moreover, key enzymes for valine degradation including Bckdha, Echs1, and Hibadh are downregulated. Also, nearly all enzymes in purine metabolism showed up-regulation in the examined datasets which led to the accumulation of urate as the end product of this pathway (Fig. 8). To provide a holistic view of the functional relationships between key dysregulated enzymes, the map of affected processes is depicted (Fig. 9).
{kind=link}
{kind=link}