2.1 Two-step induction to generate hair cell-like cells from hESCs
The hESC line X1, obtained from Prof. Xiao, College of Animal Science, Zhejiang University, was cultured on inactivated mouse embryonic fibroblast (MEF) feeder cells with DMEM/F12 supplemented with 20% knockout serum replacement (KOSR; Gibco, Shanghai, China), 1% nonessential amino acids, 2 mM L-glutamine (Invitrogen, Shanghai, China), 0.1 mM 2-mercaptoethanol (Sigma, Shanghai, China), 50 mg/ml ampicillin, and 4 ng/ml of basic fibroblast growth factor (bFGF; Invitrogen). Before induction of the differentiation, the hESCs were dissociated into small clumps using collagenase IV (Invitrogen), and these small clumps were further dissociated using 0.025% trypsin-EDTA (Sigma). The tryptic digestion was terminated using a trypsin inhibitor, and the cell suspension was passed through a 100-µm cell strainer (BD Labware, Shanghai, China) to retain few clumps of two to three cells.
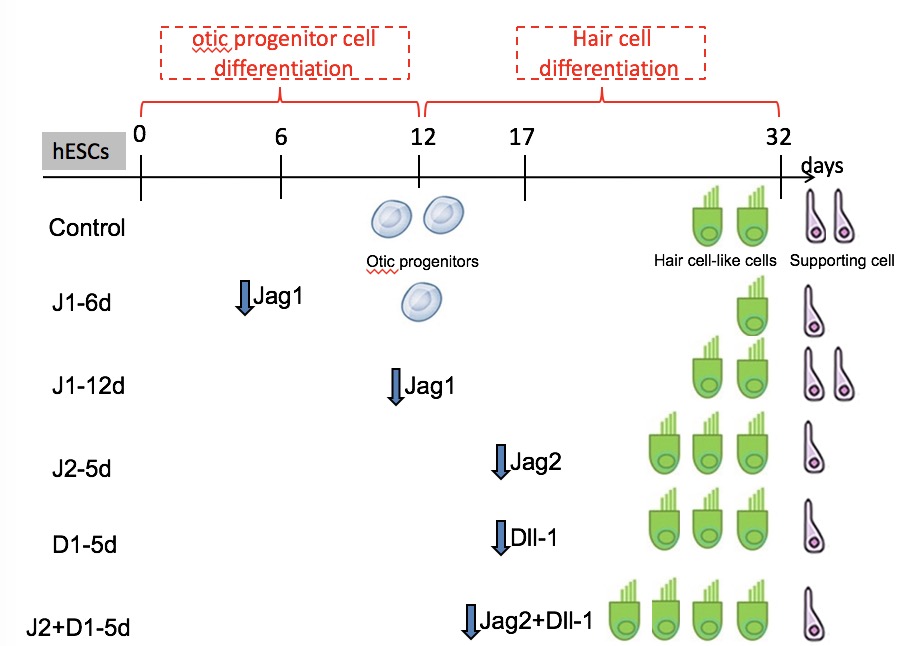
For otic progenitor differentiation, cells were plated at the density of 1 × 104 cells/cm2 on laminin-coated plastic (5 µg/cm2; R&D systems, Shanghai, China) and incubated in DMEM/F12 supplemented with N2 (1:100), B27 (1:50), FGF3 (50 ng/ml), and FGF10 (50 ng/ml (Invitrogen) for 12 days. The medium was replaced with fresh medium every two days. After 12 days of differentiation, most hESCs had differentiated into otic progenitor cells. There were two morphologically distinct types of otic colonies [18]. One cell population exhibited a flat phenotype with a large amount of cytoplasm and formed epithelioid islands; these were identified as otic epithelial progenitors (OEPs). The second population had small cells with denser chromatin, and presented cytoplasmic projections; this population was identified as otic neural progenitors (ONPs). To enrich OEPs, cells surrounding the epithelial colony were lifted by a quick incubation with Accutase (Invitrogen), at 37°C for 2–3 min. As colony edges started to curl, cells surrounding the epithelial colonies were rinsed off. A prolonged accutase treatment step permitted the collection of epithelial colonies that remained attached. For induction of OEPs to differentiate into hair cell-like cells, we used laminin (Invitrogen) as a substrate [17]. Briefly, conditioned medium was used for hair cell differentiation. The conditioned medium was from a chicken utricle stromal cell culture and was supplemented with EGF (20 ng/ml) and RA (10− 6 M). The conditioned medium was replaced every second day.
2.2 The expression pattern of genes specific for Notch signaling was analyzed during the different stages of hESC differentiation to hair cell-like cells.
Total RNA was extracted using Trizol reagent (TaKaRa, Shanghai, China) from cells in different stages of otic progenitor differentiation (otic progenitor differentiation for 6 and 12 days) and hair cell differentiation (hair cell differentiation for 5, 10, 12, 14, 16, 18, and 20 days). After reverse transcripting, the expression at mRNA level of Notch signaling pathway receptor NOTCH1 and ligands JAG1, JAG2, DLL1 were analyzed by quantitative real-time polymerase chain reaction (qRT-PCR), which was performed with LightCycler 480 using the default thermal cycling conditions (10 min at 95°C and 45 cycles of 15 s at 95°C plus 1 min at 60°C). HPRT1 and B2M were used as the endogenous control genes used for normalization. Relative quantification was performed using the comparative Ct (threshold cycle) method.
2.3 Recombinant lentiviral vector construction and cell transfection
To target different regions of each mRNA sequence (Table S1), four shRNAs targeting four different sequences of JAG-1, JAG-2, and DLL-1 (JAG-1-shRNA 1–4 for JAG-1, JAG-2-shRNA 1–4 for JAG-2, and DLL-1-shRNA 1–4 for DLL-1) and one negative control vector (NC-shRNA) were constructed by Jikai Genetics (Shanghai, China) (Table S2). Each shRNA sequence was inserted into the lentiviral vector (pAJ-U6-CMV-Puro/GFP) to construct the recombinant lentiviral vector pAJ-U6-shRNA-CMV-Puro/GFP. Subsequently, three plasmids pAJ-U6-shRNA-CMV-Puro/GFP, psPAX2 (gag/pol element), and pMD2.G (VSVG element) were transfected into 293T cells using Lipofectamine 3000 to generate Lenti-shRNA-GFP/puro viral particles. Reliable real-time PCR protocols were performed to titrate the lentivirus based on the number of proviral DNA copies present in the genomic DNA extracted from transduced cells or from vector RNA. These production and concentration methods resulted in high-titer vector preparations (Table 1). Subsequently, JAG-1-recombinant lentiviral vectors were used to infect cells on day 12 of otic progenitor differentiation. JAG-2-recombinant lentiviral vectors or DLL-1-recombinant lentiviral vectors were used to infect cells on day 5 of hair cell differentiation. After 48 h, total RNA was extracted from these cells to perform qRT-PCR to analyze gene silencing efficiency. Then, the shRNA with the highest knockout efficiency was selected, we selected lentiviral vectors harboring these shRNAs to infect cells on day 6 and 12 of otic progenitor differentiation, and on day 5 of hair cell differentiation. After 48 h of infection, the cells were collected, filtered through a 300-mesh cell strainer, and suspended in 500 µL DMEM/F12 (minus phenol red) supplemented with 10 µM Y-27632, 200 U/ml penicillin, and 200 µg/ml streptomycin. GFP-expressing cells were then sorted by flow cytometry (FCM). To unify the timing of sorting, we started the differentiation procedure at different times to ensure the time consistency of infection, collection, and sorting for all conditions. The sorted cells infected on day 6 of otic progenitor differentiation were cultured under the required conditions for an additional 6 days. Subsequently, some cells were collected for the analysis of otic progenitor differentiation, and other cells were transferred to laminin-coated plastic containing conditioned medium supplemented with EGF (20 ng /ml), RA (10− 6 M), and Y-27632 (10 µM) for 20 days of hair cell differentiation. The sorted cells infected on day 12 of otic progenitor differentiation and on day 5 of hair cell differentiation were directly transferred to laminin-coated plastic containing the conditioned medium supplemented with EGF (20 ng /ml), RA (10− 6 M), and Y-27632 (10 µM) for 15 and 20 days of hair cell differentiation. The medium was replaced with fresh medium every second day. Cells infected with lentivirus containing NC-shRNA were used as a control for analysis on the effects of each gene silence on the differentiation by qRT-PCR.
Table 1
Analysis of lentivirus titers by quantitative PCR
|
Lentivirus
|
Item
|
V Value
|
C Value
|
N Value
|
D Value
|
Viral titer
|
Mean titer
|
|
JAG-1-shRNA2
|
1
|
10
|
37
|
1×105
|
1
|
3.70E + 08
|
|
| |
2
|
1
|
3.98
|
1×105
|
1
|
3.98E + 08
|
3.70E + 08
|
| |
3
|
0.1
|
0.34
|
1×105
|
1
|
3.40E + 08
|
|
|
JAG-2-shRNA4
|
1
|
10
|
32
|
1×105
|
1
|
3.20E + 08
|
|
| |
2
|
1
|
3.52
|
1×105
|
1
|
3.52E + 08
|
3.20E + 08
|
| |
3
|
0.1
|
0.29
|
1×105
|
1
|
2.90E + 08
|
|
|
DLL-1-shRNA3
|
1
|
10
|
27.6
|
1×105
|
1
|
2.76E + 08
|
|
| |
2
|
1
|
3.05
|
1×105
|
1
|
3.05E + 08
|
2.74E + 08
|
| |
3
|
0.1
|
0.24
|
1×105
|
1
|
2.40E + 08
|
|
|
NC-shRNA
|
1
|
10
|
42
|
1×105
|
1
|
4.20E + 08
|
|
| |
2
|
1
|
4.46
|
1×105
|
1
|
4.46E + 08
|
4.19E + 08
|
| |
3
|
0.1
|
0.39
|
1×105
|
1
|
3.90E + 08
|
|
2.4 Western blot
Cells were washed twice with ice-cold PBS and protein was extracted. The extract was centrifuged at 12,000 rpm for 15 min at 4°C to remove cellular debris. Protein concentrations were determined by the Bradford method where in 20 µg of protein sample was heated to 95°C for 5 min, run on 10% SDS-PAGE gel, and transferred to PVDF membrane (Millipore, Shanghai, China) using the semidry transfer method. The membranes were blocked for 1 h in Tris-buffered saline containing 0.01% Tween 20 with 10% non-fat dried milk and incubated overnight at 4°C with the relevant antibodies including anti-DLL-1, anti-JAG-1, and anti-JAG-2 (Santa Cruz Biotechnology, Shanghai, China). After washing, the membranes were incubated with a peroxidase-conjugated anti-IgG secondary antibody, (Bio-Rad Laboratories, Shanghai, China) for 1 h. Protein bands were detected using an enhanced chemiluminescence detection kit. All the bands were analyzed using Image J software (version 1.6 NIH) to determine the relative levels of Notch signaling ligands compared to β-actin expression.
2.5 Gene expression specific analysis
Total RNA was extracted and reverse transcribed (Fermentas, Shanghai, China). The cDNA was used as a template for PCR using the primer pairs listed in Table 2. All RT-PCR results presented were confirmed by at least two independent controlled experiments. PCR products were electrophoresed on a 1.2% agarose gel, and stained bands were visualized under UV light and photographed. Band intensities were analyzed quantitatively in triplicate using Image J software, and the expression level of each gene was normalized to that of GAPDH.
Table 2
Primers of RT-PCR for marker genes specific for otic progenitors and hair cells
|
Gene
|
Primer
|
Sequence
|
Tm
|
|
Sox2
|
Sense
|
5’- GGG AAA TGG GAG GGG TGC AAA AGA GG -3’
|
57℃
|
|
Antisense
|
5’- TTG CGT GAG TGT GGA TGG GAT TGG TG -3’
|
|
Oct4
|
Sense
|
5’- GAC AGG GGG AGG GGA GGA GCT AGG − 3’
|
58℃
|
|
Antisense
|
5’- CCT CCC TCC AAC CAG TTG CCC CAA AC -3’
|
|
Nanog
|
Sense
|
5’- ACC TAT GCC TGT GAT TTG − 3’
|
60℃
|
|
Antisense
|
5’- AGA AGT GGG TTG TTT GC -3’
|
|
Pax8
|
Sense
|
5’- ACC CCC AAG GTG GTG GAG AAG A -3’
|
62℃
|
|
Antisense
|
5’- CTC GAG GTG GTG CTG GCT GAA G -3’
|
|
Pax2
|
Sense
|
5’- GAG CGA GTT CTC CGG CAA C -3’
|
60℃
|
|
Antisense
|
5’- GTC AGA CGG GGA CGA TGT G -3’
|
|
Six1
|
Sense
|
5’- GAC TCC GGT TTT CGC CTT TG -3’
|
57℃
|
|
Antisense
|
5’- TAG TTT GAG CTC CTG GCG TG -3’
|
|
GATA3
|
Sense
|
5’-GTA CAG CTC CGG ACT CTT CCC-3’
|
60℃
|
|
Antisense
|
5’- CTG CTC TCC TGG CTG CAG ACA − 3’
|
|
Dlx5
|
Sense
|
5’- TTC CAA GCT CCG TTC CAG AC -3’
|
57℃
|
|
Antisense
|
5’- GTA ATG CGG CCA GCT GAA AG -3’
|
|
Eya-1
|
Sense
|
5’- TCA GAT GCT ATC TGC CGC TG -3’
|
57℃
|
|
Antisense
|
5’- GTG CCA TTG GGA GTC ATG GA -3’
|
|
Atoh1
|
Sense
|
5’- GCC GCC CAG TAT TTG CTA CA -3’
|
57℃
|
|
Antisense
|
5’- GCT AGC CGT CTC TGC TTC TG -3’
|
|
Myosin7A
|
Sense
|
5’- CAC ATC TTT GCC ATT GCT GAC − 3’
|
55℃
|
|
Antisense
|
5’- AGA AGA GAA CCT CAC AGG CAT − 3’
|
|
Espin
|
Sense
|
5’ - CAG GCA TGT CCT CAC CCA AT -3’
|
55℃
|
|
Antisense
|
5’- CGT GGC GGA GTT TGT TCT TG -3’
|
|
Brn3c
|
Sense
|
5’- TGC AAG AAC CCA AAT TCT CC -3’
|
55℃
|
|
Antisense
|
5’- GAG CTC TGG CTT GCT GTT CT -3’
|
|
P27kip1
|
Sense
|
5’- CTG GAG CGG ATG GAC GCC AGA C -3’
|
62℃
|
|
Antisense
|
5’- CGT CTG CTC CAC AGT GCC AGC − 3’
|
|
GAPDH
|
Sense
|
5’- GAA GGT CGG AGT CAA CGG − 3’
|
58℃
|
|
Antisense
|
5’- GGA AGA TGG TGA TGG GAT T-3’
|
2.6 Immunocytochemistry
Cells were fixed with 4% paraformaldehyde for 15 min and permeabilized with PBS containing 0.25% Triton X-100 and 5% normal donkey serum for 10 min at room temperature. Blocking was performed in PBS containing 1% bovine serum albumin and 0.1% Tween-20, which was followed by three washes of 5 min each with PBS. The cells were incubated with primary antibody overnight at 4°C. After washing, Specific antibody binding was visualized with donkey anti-mouse, anti-goat, or anti-rabbit secondary antibodies conjugated to either Alexa Fluor 488 or Alexa Fluor 594 (Jackson, Shanghai, China). The dilution ratio of all antibodies was based on the reference [17]. Nuclei were visualized with DAPI (4',6-diamidino-2-phenylindole). Images were acquired with a Zeiss Axiophot fluorescence and confocal microscope.
2.7 SEM assay
Cells differentiated for 3 weeks were fixed overnight in 2.5% glutaraldehyde (Sigma) at 4°C. The specimens were washed twice with PBS for 20 min each and treated with 1% osmium tetroxide (Sigma) for 30 min. The specimens were washed with PBS for 10 min and dehydrated in a graded ethanol series. Thereafter, ethanol was replaced with isoamyl acetate (Aladdin, Shanghai, China) for 20–30 min, and the specimens were dried using critical point drying method. The specimens were viewed with a Hitachi S-3000N variable pressure SEM.
2.8 Statistical analysis
Data are presented as mean values ± standard deviation (SD) with the number of independent experiments (n) indicated. All collected data were examined by multifactorial analysis of variance. Statistical differences between the independent variables were assessed by post hoc tests (Tukey’s studentized range tests for variables). All tests were two-tailed, and statistical significance was set at P < 0.05.
{kind=link}