Sequencing and mining of potential SNPs in lotus
A total of 138 F2 individuals were developed from a cross between ‘BG’ and ‘WR1’. Two parents were sequenced at a relatively high coverage using WGS. In total, 10.8 Gb (11.6-fold genome coverage) and 9.9 Gb (10.7-fold genome coverage) high-quality short-read sequences were generated for ‘BG’ and ‘WR1’, respectively (Table S2). All high-quality reads from ‘BG’ and ‘WR1’ were aligned to the repeat masked reference genome of ‘China Antique’ to detect putative SNPs. As results, 70.5 % and 85.4 % of the reads (Q > 20) from ‘BG’ and ‘WR1’ were aligned to the reference genome. A total of 949,840 and 900,010 putative SNPs were identified in ‘BG’ and ‘WR1’, respectively (Table 1). Among them, 296,706 SNPs were polymorphic between the two parents. For the polymorphic SNPs, 244,926 and 44,802 were heterozygous for ‘BG’ and ‘WR1’, respectively, indicating a high rate of heterozygosity in the maternal variety. Polymorphic SNPs markers were grouped as, aaxbb, lmxll, nnxnp, and hkxhk, based on parental genotypes, each with 27,738, 224,166, 24,042, and 20,760 markers, respectively (Table 1).
Table 1
Number of SNP detected in the parents and F2 individuals
| Marker type | Number of SNP in 'BG' | Number of SNP in 'WR1' | Number of SNP in F2 | Number of bin in F2 |
| Total | 949840 | 900010 | | |
| Missing marker | 155924 | 205754 | | |
| Non polymorphic marker between the parents | 447380 | | | |
| Polymorphic marker between the parents | 296706 | | 285895 | 4031 |
| aaxbb | 27738 | | 27527 | 1069 |
| lmxll | 224166 | | 214643 | 1933 |
| nnxnp | 24042 | | 23811 | 638 |
| hkxhk | 20760 | | 19914 | 391 |
| Homozygous SNP | 51780 | 251904 | 27527 | |
| Heterozygous SNP | 244926 | 44802 | 258368 | |
An overall genome sequence of 482.6 Gb was generated in the 138 F2 individuals, with an average of 5.3 Gb for each individual, which was equivalent to approximately 5.7× coverage of the reference genome (Table S2). As a result, 64.2% of high-quality reads (Q > 20) of the F2 individuals was mapped on the reference genome. Based on the parental polymorphic SNPs, a total of 285,895 SNPs were identified, with 27,527, 214,643, 23,811, and 19,914 in aaxbb, lmxll, nnxnp, and hkxhk, respectively. Moreover, these SNPs were binned as 1,069, 1,933, 638, and 391 markers in aaxbb, lmxll, nnxnp, and hkxhk, respectively (Table 1).
Construction of linkage map and comparison with the draft genome
Due to high heterozygosity of both parental genomes, most loci in the F2 progenies were heterozygous. The hkxhk marker could not be distinguished from the parents, thus it was not used for genetic linkage map construction. As a result, the bins from aaxbb, lmxll, and nnxnp SNP were merged to construct genetic linkage map (Tables 2 and S3). This genetic map contained 2,935 bins generated from 236,840 SNPs. These bins were distributed on eight genetic groups spanning 896.11 cM with an average marker interval of 0.27 cM. Marker distribution and linkage length varied among linkage groups. LG1 was the largest with 170.99 cM, and contained 617 markers. The shortest group LG8 was 33.30 cM, and contained 114 markers. The highest marker density was on LG6, with an average marker interval of 0.21 cM. The lowest marker density was on LG4, with an average marker interval of 0.47 cM. The largest gap among all linkage groups was 9.63 cM on LG7. The information of all markers including marker name, linkage group, genetic distance, and marker type are listed in Table S3.
Table 2
Summary of genetic linkage map
| Linkage group | No. of SNP | No. of bin | Length (cM) | Average distance (cM) | Max. gap (cM) |
| LG1 | 55633 | 617 | 170.99 | 0.28 | 7.24 |
| LG2 | 44251 | 567 | 161.53 | 0.28 | 9.55 |
| LG3 | 28368 | 292 | 120.38 | 0.41 | 5.97 |
| LG4 | 12997 | 247 | 117.07 | 0.47 | 5.89 |
| LG5 | 35287 | 356 | 112.08 | 0.31 | 8.63 |
| LG6 | 27924 | 520 | 106.64 | 0.21 | 7.10 |
| LG7 | 14917 | 222 | 74.12 | 0.33 | 9.63 |
| LG8 | 17463 | 114 | 33.30 | 0.30 | 4.61 |
| Total | 236840 | 2935 | 896.11 | | |
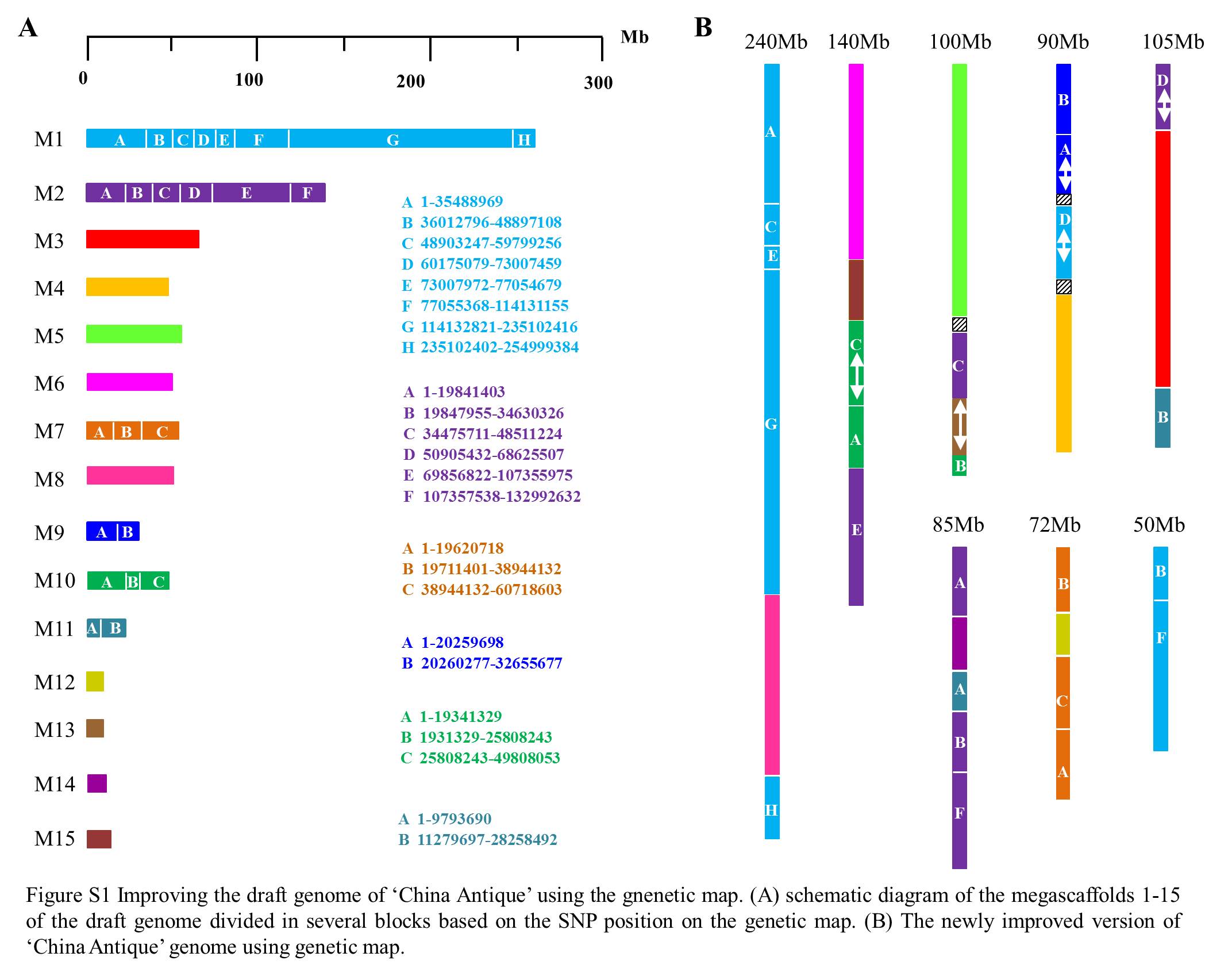
The genetic map was compared with the lotus draft genome (Fig. 2). The eight linkage groups were integrated on megascaffolds 1 to 15. All linkage groups except LG8 were anchored to at least two megascaffolds. LG1 covered the 1–35 Mb, 49–60 Mb, 73–77 Mb, 114–255 Mb regions of megascaffolds 1 and 8, respectivley. LG2 was anchored to the 70–110 Mb region of megascaffolds 2 and 6, and to 1–20 Mb or 26–50 Mb region on megascaffolds 10 and 15, respectively. LG3 covered the 34–48 Mb region of megascaffolds 2 and 5, 20–25 Mb region of megascaffolds 10, and 13. LG4 covered the 60–73 Mb region of megascaffolds 1, 4 and 9. LG5 covered the 50–69 Mb region of megascaffolds 2 and 3, and 11–28 Mb region of megascaffold 11. LG6 was anchored to the 1–35 Mb and 107–133 Mb regions of megascaffold 2 and to 1–10 Mb region of megascaffolds 11, and 14. LG7 was anchored to megascaffolds 7 and 12. Therefore, megascaffolds 1, 2, 7, 9, 10, and 11 were divided into several blocks, and resettled with megascaffolds 3, 4, 5, 6, 8, 12, 13 14 and 15. Taken together, the original 15 megascaffolds in the draft genome were integrated into novel 8 megascaffolds (Figure S1).
QTL analysis for rhizome enlargement
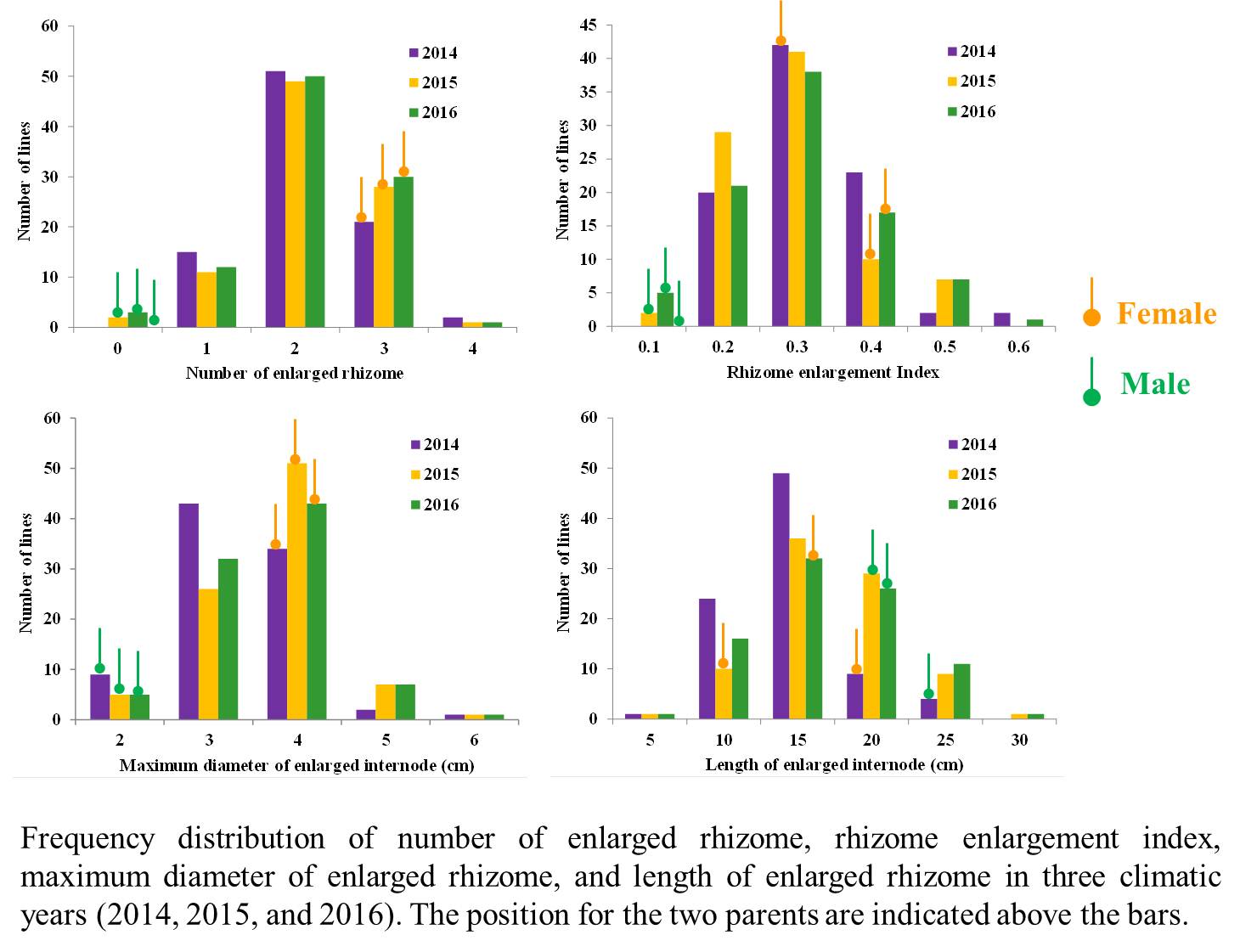
The variation in phenotypic traits among segregating population was used as a key parameter for QTL mapping. Four phenotypic traits, NER, MDE, LER and REI, were distributed continuously in the F2 population. Significant transgressive segregation was observed for each trait (Figure S2). ANOVA results showed that genotype, environment and their interactions had highly significant effects (P < 0.001) on these traits, indicating that the three traits were easily influenced by environmental factors (Table S4).
Mapping analysis for NER and REI identified 14 significant QTLs in the F2 population in three climatic years (Table 3, Fig. 3A). For NER, four QTLs distributed on LG5 and LG6 were detected, with their phenotypic variation ranging from 7.5 to 14.8%. The region which covered 9.4–11.8 cM on LG6 was repeatedly found across the three years. Ten QTLs on LG1, LG2, LG5 and LG6 were identified for REI in the three years, with phenotypic variation of these QTLs ranging from 6.6 to 22.8%. A region covering 28.4–29.9 cM on LG1 was consistently detected in two years, and another region covering 12.3–13.5 cM on LG1 was detected in all the three climatic years (Table 3).
Table 3
Significant QTLs identified for the traits in three climatic years
| Trait | Name of QTL | Year | LG | Peak a | C.I.b | LOD c | R2 d | Additive e |
| Number of enlarged rhizome | qNER-LG6-14 | 2014 | 6 | 10 | 9.4–11.8 | 2.73 | 7.45 | 0.35 |
| | qNER-LG6-15 | 2015 | 6 | 10 | 9.4–11.8 | 2.82 | 11.53 | 0.31 |
| | qNER-LG8-15 | | 5 | 29 | 28.0-29.7 | 3.94 | 13.59 | -0.38 |
| | qNER-LG6-16 | 2016 | 6 | 10 | 8.6–11.8 | 3.27 | 14.85 | 0.39 |
| Rhizome enlargement Index | qREI-LG1-14 | 2014 | 1 | 29 | 28.4–29.9 | 4.09 | 11.48 | 0.05 |
| | qREI-LG2-14a | | 2 | 52 | 51.3–53.5 | 3.45 | 8.75 | 0.03 |
| | qREI-LG2-14b | | 2 | 12 | 11.6–13.5 | 3.61 | 20.00 | 0.05 |
| | qREI-LG6-14 | | 6 | 10 | 9.5–10.5 | 3.44 | 9.55 | 0.05 |
| | qREI-LG5-14 | | 5 | 29 | 28.0-29.7 | 4.98 | 14.52 | -0.05 |
| | qREI-LG1-15 | 2015 | 1 | 29 | 28.4–29.9 | 2.65 | 10.91 | 0.03 |
| | qREI-LG2-15 | | 2 | 13 | 12.3–14.0 | 2.72 | 22.80 | 0.04 |
| | qREI-LG1-16a | 2016 | 1 | 162 | 160.9-165.8 | 2.53 | 9.14 | 0.03 |
| | qREI-LG1-16b | | 1 | 46 | 45.9–47.1 | 2.84 | 6.67 | 0.03 |
| | qREI-LG2-16 | | 2 | 12 | 11.6–13.5 | 3.65 | 19.96 | 0.06 |
| a The peak position of QTL; b Confidence interval at P = 0.05; c LOD score as calculated by WinQTLCart 2.5. d Represents the phenotypic variation accounted for; |
| e Additive effect, positive value (+) means the allele is derived from the female parent ‘BG’, negative value (−) means the allele is derived from the male parent ‘WR1’. |
These significant QTLs with overlapping confidence intervals were integrated into the consensus QTLs (Fig. 3A). As a result, one consensus NER QTL designated cqNER-LG1, with a positive additive effect and covering 2.4 cM, was integrated. Two consensus QTLs, cqREI-LG1 and cqREI-LG2, were identified for REI covering a 1.5 cM and 1.2 cM regions on LG1 and LG2, respectively. cqREI-LG2 explained about 20% of the phenotypic variation, and was the major QTL for REI. The two consensus REI QTLs had a positive additive effect, suggesting that alleles from the female parent of ‘BG’ contributes to rhizome enlargement.
Candidate genes underlying the major cqREI-LG2 QTL interval
A major cqREI-LG2 QTL showing 20% of the phenotypic variation and with a rhizome-enlargement effect were identified across the three-year QTL mapping period. The QTL interval of cqREI-LG2 covered a 4.5 Mb region on megascaffold 6, which harbored 59 annotated genes (Table S5). Of these genes, 11 had low or no expression in all tested tissues including leaves, petiole, petals, pistils, stamens, roots and rhizomes, while the remaining 48 genes were highly expressed in most tissues (Figure S3). A previous transcriptome data showed an increase in the number of candidate genes controlling the conversion of stolon to rhizome between the ZT1 and ZT2 or RT2 and RT3 developmental stages in two lotus cultivars of ‘RL’ (tropical lotus with stolon) and ‘ZO’ (temperate lotus with enlarged rhizome), and these genes were considered candidate for rhizome girth enlargement (Yang et al. 2015). Of the 48 highly expressed genes in this study, nine were predicted as candidate genes for rhizome girth enlargement (Fig. 3B). Among them, four including NNU_05272, NNU_05296, NNU_05305 and NNU_05314 involved in cell and liqlid metabolism were observed. A soluble starch synthase-like gene, NNU_05273, was also identified. In addition, two genes, NNU_05287 and NNU_05299, encoding ubiquitin E2 enzyme and peroxidase, respectively, were detected. Moreover, a BEL1-like homeobox protein 6 homolog gene, NNU_05289, and a bHLH transcription factor, NNU_05313, were identified. Expression levels of NNU_05273, NNU_05289 and NNU_05305 increased at the RT3 and ZT2, while the remaining six genes decreasd at these two stages (Fig. 3B).
StBEL5 has been shown to be a transcriptional activator of StSP6A which acts as a tuberization signal in potato (Abelenda et al. 2014; Sharma et al. 2016). The BEL1-like homeobox protein 6 homolog gene, NNU_05289, designated as NnBEL6, is most likely candidate gene that potentially control rhizome enlargement. Thus we sequenced 8,485bp of the genomic region of NnBEL6 from 1.5 kb upstream to the stop codon in the two parental genotypes and other 23 lotus accessions phenotypically classified into class I, II and III with varying REI as mentioned above (Fig. 3C). A total of 17 polymorphic sites were identified, with four in the promoter region, one in the first exon (+ 124), ten in the first intron, one in the second intron (5,923) and one in the fourth exon (6,695). Only two polymorphic sites at -1,347 and 3,741 were detected between class I and II lotus. The remaining 15 polymorphic sites were observed between class III lotus and those of class I or II, and were highly conserved in most varieties, except for two lotus accessions (Fig. 3C). These results suggested that two polymorphic sites at -1347 and 3741 in NnBEL6 could be crucial for determining rhizome phenotype in lotus. Thus, NnBEL6 is a key candidate gene regulating rhizome enlargement in lotus.
Functional validation of NnBEL6 gene in promoting rhizome enlargement
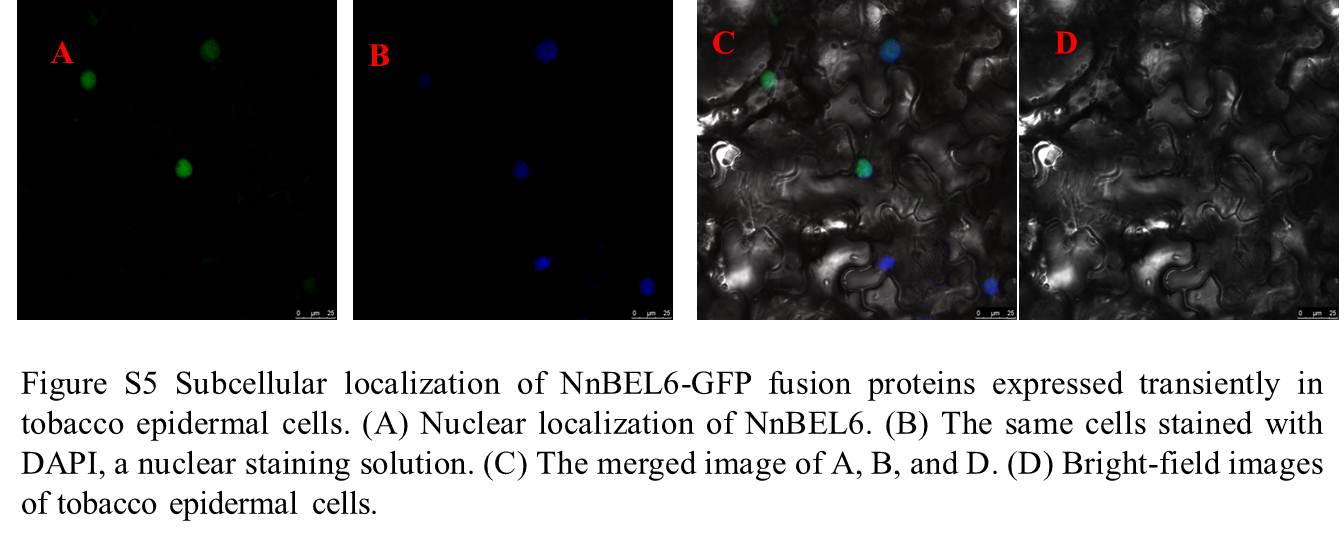
NnBEL6 encodes a putative protein of 751 amino acids and contains four conserved regions: the conserved BELL domain (Ger340-Ile424), the homeodomain (Trp393-Glu540), the N-terminal SKY, and C-terminal VSLTLGL boxes (Figure S4A). Phylogenetic analysis constructed with the alignments of NnBEL6 protein with the BEL-like genes from Arabidopsis thaliana and Solanum tuberosum revealed a close relationship between NnBEL6 and AtBEL6 or AtBEL7 (Figure S4B). The tissue distribution and expression pattern of NnBEL6 were determined by qRT-PCR analyses (Fig. 4). NnBEL6 was expressed in the leaf, petiole, flowerbud, pedicel, rhizome and root at the stolon and initial swelling stage of ‘ZO’ (Fig. 4B). At the stolon stage, NnBEL6 transcript was highly accumulated in the rhizome and roots, but with decreased levels in the leaf, petiole, flower, and pedicel. At the initial swelling stage, NnBEL6 was primarily expressed in the rhizome with a four-fold higher transcript levels than that at the stolon stage. In contrast, the expression of NnBEL6 in other tested tissues showed no significant variation. NnBEL6 expression in first rhizome internode was higher in ‘ZO’ than that in ‘WR1’ at the initial swelling stage (Fig. 4C). Comparison of rhizome change and NnBEL6 mRNA levels over the developmental time-course indicated its strict correlation with the developmental process, suggesting that NnBEL6 might play a positive role in controlling the initial rhizome enlargement. Subcellular localization of NnBEL6 performed in tobacco epidermal cells indicated that NnBEL6 was a nuclear protein (Figure S5).
To further confirm the function of NnBEL6 in the formation of storage organ, an overexpressing NnBEL6 construct driven by the CaMV 35S promoter was transformed into the potato cultivar ‘E3’, which can only form microtuber under SD in vitro (Fig. 5). The results demonstrated that under SD, overexpressing NnBEL6 promoted tuber formation. The transgenic lines developed microtuber at approximately 95 days after transplantation (DAT), while microtuber formation started in the WT ‘E3’ controls at 109 DATs (Figs. 5A and B). The transgenic lines formed an average of 3.2 microtubers per plant at 120 DATs (Fig. 5C). To dissect the effect of NnBEL6 on signal transduction in the photoperiodic tuberization pathway, we further assessed the endogenous expression of two reported key genes in the photoperiodic tuberization pathway, StSP6A and StSP5G, in the transgenic lines. The expression level of StSP6A was significantly up-regulated in 35S:NnBEL6 transgenic plants (Fig. 5D), which was consistent with the observed early tuberization result. However, the expression level of StSP5G showed varied profiles, with a slight down-regulation in two transgenic plants, and an up-regulation in one transgenic plant (Fig. 5E).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}