Reagents
Mouse macrophage RAW264.7 cell line and human embryonic kidney 293 (HEK293) cell line were obtained from the American Type Culture Collection (Rockville, MD, USA). Cells were cultured with Alpha minimal essential Medium (α-MEM; Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS; Gibco, Grand Island, NY, USA) and 1% penicillin-streptomycin (Hyclone, Thermo Scientific, MA, USA) in a 37℃ incubator with 5% CO2. Recombinant mouse RANKL and M-CSF were purchased from R&D Systems (Minneapolis, MN, USA). Alizarin stain kit were obtained from Solarbio Life Sciences (Beijing, China). Antibodies against Histone 3 (bs-17422R), CD81 (bs-2489R), TSG101 (bs-1365R), LaminA/C (bs-1839R), Osteocalcin (bs-0470R), LC3 (bs-8878R), SQSTM1/P62 (bs-2951R) and FAM134A (bs-14725R) and β-actin (bs-0061R) were purchased from Bioss Antibodies (Beijing, China).
Isolations of BMMs and BMSCs
In present study, BMMs were used studying osteoclast differentiation and BMSCs were used investigating osteogenic differentiation. The detailed processes of BMMs and BMSCs separation have been described in our previous studies [28, 29]. Briefly, 8 week old male C57BL/6 mice were euthanatized and mouse femur and tibia were dissected under aseptic conditions. The medullary cavity of tibia and femur were washed with α-MEM to collect the bone marrow cells (BMCs). After BMCs were cultured in α-MEM for 24 hours, the non-adherent cells were removed through renewing complete medium (α-MEM, 10% FBS, 1% penicillin-streptomycin) to obtain BMSCs. As for BMMs generation, collected BMCs were stimulated with 50 ng/ml M-CSF for 96 hours to obtain BMMs.
Osteoclast differentiation and TRAP staining
To generate osteoclasts,5 × 103 BMMs were seeded in 96-well plates and cultured with α-MEM which contains 50 ng/ml M-CSF, 100 ng/ml RANKL 10% FBS and 1% penicillin-streptomycin for 96 hours. For TRAP staining, culture medium was removed and cells were washed three times with phosphate-buffered saline (PBS, Hyclone, Thermo Scientific, MA, USA) and fixed in 4% paraformaldehyde (PFA, Biosharp, Anhui, China) for 20 mins. PFA were removed and cells were washed with PBS one more time. Subsequently, cells were stained with TRAP staining solution (Sigma-aldrich, Shanghai, China) for 1 hour. TRAP assay kit (Beyotime Biotechnology, Jiangsu, China) was used to detect relative TRAP activity according instruction manual.
Separation of OC-sEVs
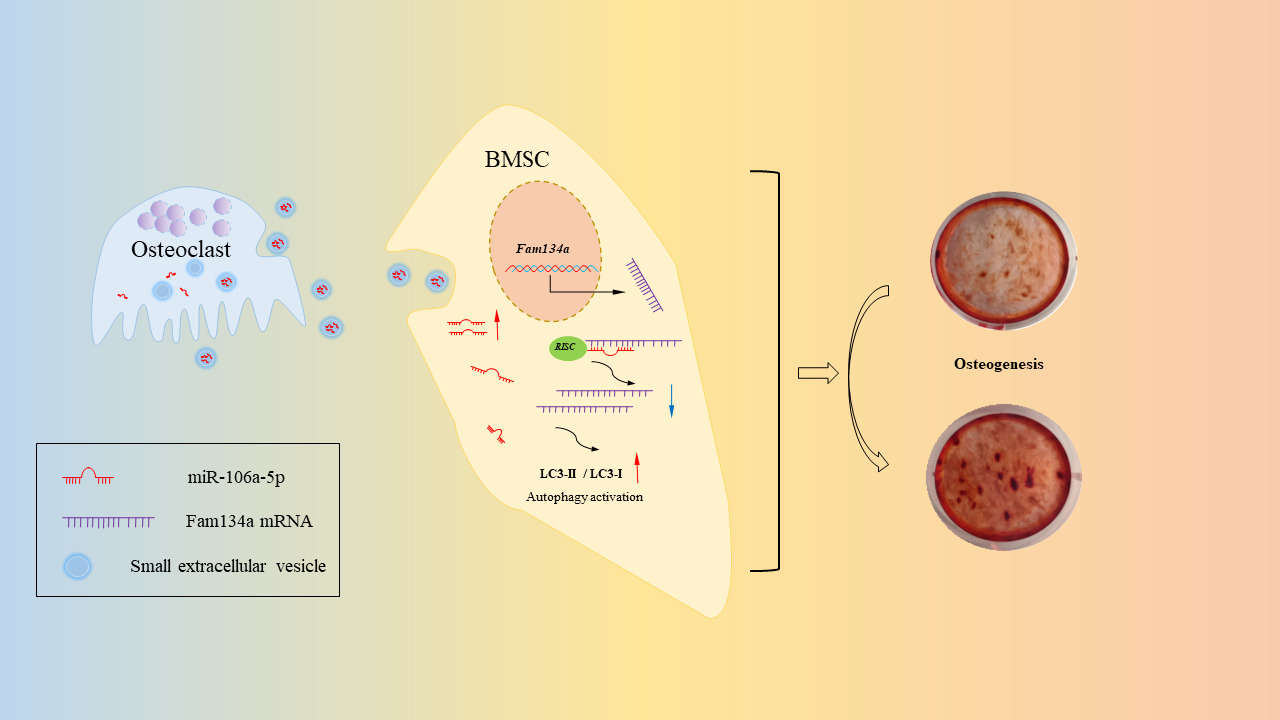
Osteoclast-derived sEVs were collected through centrifugation with a series of speed gradients as previously described with some modifications [30]. Briefly, culture medium was removed and osteoclasts were washed three times with PBS. For sEVs isolation, osteoclasts were cultured in a-MEM for 24 hours to get supernatant. To remove the dead cells and apoptotic bodies, the supernatant was centrifuged at 1,500 × g for 15 mins and 3,000 × g for 15 mins. To remove lEVs, the supernatant was centrifuged at 18,000 × g for 30 mins. Finally, sEVs were pelleted via centrifugation at 110 000 × g for 70 mins. The pellets were washed in 50 ml PBS and the centrifuged again at 110 000 × g for 70 mins. The separated sEV were resuspended in PBS for experiments. All centrifugation operations were performed at 4°C using Optima XE-90 (Beckman Coulter).
Characterization of OC-sEVs
For transmission electron microscopy (TEM) analysis, the 20 μl separated sEVs were dropped on each formvar copper grid and fixed with 20 μl 4% PFA. After fixation, the liquid on the surface of copper grids were removed by absorbent paper and uranyl acetate (Electron Microscopy Sciences) were dropped on formvar copper grids for staining. After 10 mins at room temperature, uranyl acetate was removed and formvar copper grids were naturally dried for observation. Digital images were obtained under FEI Tecnai microscope at an accelerating voltage of 80 kV. For nanoparticle tracking analysis (NTA) analysis, the size distribution and concentration of separated sEVs was examined using a ZataView (Particle 140 Metrix, Germany).
miRNA microarray analysis
The sEVs secreted by osteoclasts at each differentiation stages were isolated by ultracentrifugation. miRNeasy Mini Kit (Qiagen) was used to extract total RNA from the isolated sEVs and purity of RNAs were examined by Agilent 2100 Bioanalyzer before microarray analysis. Mouse miRNA Microarray Kit (Agilent, V3) was used and we added 100 ng total RNA in each sample. According to manufacturer's instructions, the RNA in samples were first phosphorylated and treated with cyanine 3-cytidine biphosphate. Subsequently, the samples were vacuum dried for 2 hours at 50°C and hybridized to microarray surface for 20 hours at 55°C on a shaker with 20 RPM. The glass slides then were washed and scanned with Agilent Microarray Scanner (Agilent Technologies). Feature Extraction software v11.0.1.1 (Agilent Technologies) was used to extract data and GeneSpring GX software (Agilent Technologies) was used to normalize and analyze. We used R v3.6.0 to convert data into heatmap.
Osteogenic differentiation
For osteogenic differentiation, 1 × 106 BMSCs were seeded in 24-well plates and 5 × 106 BMSCs were seeded in 6-well plates. When BMSCs covered all the bottoms of each well and stopped proliferating, osteogenic induction was performed. The osteogenic medium contains 2mM β-glycerophosphate (Aladdin, Shanghai, China), 100μM ascorbic acid (Aladdin, Shanghai, China) and 10 nM dexamethasone (Aladdin, Shanghai, China). In order to generate sEVs-contained induction medium, 1 × 109 OC-sEVs were added to per ml osteogenic medium. Subsequently, BMSCs in 24-well plates were cultured with sEVs-contained osteogenic medium for 7 days to extract total RNA. BMSCs in 6-well plates were cultured with sEVs-contained osteogenic medium for 14 days to extract total protein. Total protein and RNA extracted from BMSCs were used to assess the protein and mRNA expression of osteogenic markers by western blots and qPCR analysis. For detecting ALP activity and assessing mineralization, BMSCs cultured with sEVs-contained osteogenic medium for 14 days in 24-well plates were washed by PBS three times and fixed in 4% PFA for 30 mins. Alkaline phosphatase assay kit (Beyotime Biotechnology) and 1% Alizarin Red-S (Solarbio Life Sciences) were used to detect ALP activity and assess mineralization according to manufacturer's protocols.
Generation of sEV-modified DBM scaffolds
Decalcified bone matrix (DBM) was prepared in our study based on demineralized bovine limbs as previously reported [31]. we cut DBM into 2.5 mm cubes to fill the calvarial defect. Before sEVs coated, the DBM were immersed in 75% ethanol for 2 hours,washed three times with PBS and then incubated with 10 μg/mL fibronectin (Sigma, Shanghai, China) overnight at 37 °C. The DBM was immersed in sEV-containing medium (50μg sEVs suspended in 50μL a-MEM) and incubated at 37°C for 6 hours. Subsequently, The DBM was taken out and stored at -80°C.
Animal experiments
We evaluated the bone regeneration potency of sEV-modified DBM using a calvarial defect mouse model. After 8 week old male C57BL/6 mice were anesthetized by intraperitoneal injection of pentobarbital (Sigma Aldrich, 4mg/100g), a sagittal median incision of 1.5 cm was made and a 2.5 mm-sized defect was created on the right side of the calvarial bone using a dental micro-drill. Afterwards, the sEV-modified scaffolds were implanted into the bone defects. After 4 weeks, mice were euthanized according to the AVMA guidelines for the euthanasia of animals. the calvarial bones of mice were removed for subsequent experiments. All mice were obtained and fed in the animal facility of Third Military Medical University. The Institutional Animal Care and Use Committee of the Third Military Medical University reviewed and approved all experimental protocols.
Histological TRAP and IHC evaluation
Calvarial bones were collected at the euthanized time and fixed in 4% PFA, washed three times and decalcified in 10% EDTA solution for 2 weeks. Decalcification solution was renewed every two days. After dehydrated by ethanol gradient, decalcified calvarial bones were embedded in paraffin and sectioned into 5 μm by microtome for subsequent TRAP and IHC staining. For histological TRAP stain, sections were incubated with TRAP stain solution (Sigma-aldrich, Shanghai, China) according to the manufacturers’ instructions. The expressions of osteocalcin and FAM134A were detected by IHC assessment. Firstly, sections were incubated with primary antibodies of osteocalcin (1:300) and FAM134A (1:300) at 4°C overnight. Afterwards, EnVision+ system HRP kit (Dako, Sweden) containing biotinylated secondary antibody was used to develop color and nuclei were counterstained with hematoxylin. Finally, the washed sections were taken using an optical microscope. The German semi-quantitative scoring system was used to analysis slice data, which was described particularly in our previous studies [28].
Luciferase reporter assay
1 × 105 cells were seeded and cultured into 24-well plates until reaching 70% confluence. Lipofectamine 2000 Reagent (Invitrogen) was used to transfected 1.5 μg constructed pGL3-basic luciferase reporter plasmid per well (Promega) or 1.5 μg control luciferase plasmid (Promega) into the cells with 0.15 μg pRL-SV40 plasmid (Promega) according manufacturer's instructions. Dual-luciferase reporter assay system (Promega, USA) was used to detect luciferase and Renilla activities of transduced cells after 48 hours.
Western blot analysis
Cells or sEVs were lysed in RIPA lysis buffer mixing with protease phosphatase inhibitor (Beyotime Biotechnology, Jiangsu, China). The lysis system was incubated on ice for 30 mins, and ultrasonic lysed every 10 mins. Subsequently, the supernatant was carefully removed and transferred into a new microfuge tube after centrifugation at 13000 g for 15mins. The BCA protein assay kit (Beyotime Biotechnology, Jiangsu, China) was used to detect the concentration of proteins according manufacturer's instructions. Each sample (50 μg) was diluted in loading buffer (Beyotime Biotechnology, Jiangsu, China) and subjected to a standard SDS-PAGE followed by transferred onto polyvinylidene fluoride membranes (ImmobilonTM-PSQ Membranes, Sigma-Aldrich, China). After blocking in 5% skim milk and washing in Tris Buffered Saline Tween buffer, proteins were detected using the following antibodies: Histone 3 (bs-17422R) at a 1:1000 dilution, CD81 (bs-2489R) at a 1:1000 dilution, TSG101 (bs-1365R) at a 1:1000 dilution, LaminA/C (bs-1839R) at a 1:1000 dilution, and β-actin (bs-0061R) at a 1:2000 dilution. Corresponding secondary antibodies against primary antibodies were used by an hour of incubation (1:2000). Blots against β-actin served as loading control. Super ECL Plus (Bioground, Chongqin, China) and Immun-Star HRP (BioRad) were used to detect chemiluminescent signals.
qPCR analysis
Total RNAs from cells or sEVs were extracted using Trizol reagent (Life Technologies, NY, USA). The ultra-low volume spectrometer (BioDrop µLite, Cambridge, England) was used to measure concentrations and purity of the RNA samples by detecting OD260 and OD280. PrimeScriptTMRT reagent kit (Takara, Nojihigashi, Japan) was used to synthesize cDNA. SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) and specific primers (listed in supplement table 1) were used to further amplification process. Real-time PCR process was carried out on CFX96™ Real-Tim PCR System instrument (Bio-Rad).
Lentivirus construction and transfection
In our study, mimics and inhibitors of miR-106a-5p were purchased from GenePharma (Shanghai, China). The constructed lentivirus vectors which express miR-106a-5p or inhibit miR-106a-5p were obtained from Genechem (Shanghai, China). We logined in the National Center for Biotechnology Information and used BLAST searches (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to ensure inhibitors and products of inhibitory vector only targeted miR-106a-5p. Before transfection, 1 × 106 cells were seeded and cultured in 6-well plates until reaching 70% confluence. Complete medium was replaced to transfection medium containing a-MEM, 10% FBS and lentivirus vectors with 20 MOI (Multiplicity of Infection). After incubation for 48 hours at 37°C, the transfection medium was replaced to selection medium containing a-MEM, 10% FBS and 5 mg/ml puromycin for obtaining stable transduced cells. For rescue experiments, Lipofectamine 2000 Reagent (Thermo Scientific, MA, USA) was used to transfect Fam134a expressing plasmids (GeneCopoeia) to cells which were cultured with miR-106a-5p-sEVs or stably expressed miR-106a-5p. Exo-Fect Exosome Transfection Kit (System Biosciences) was used to load mimics or inhibitors into OC-sEVs. miR-106a-5p mimic (5’ to 3’): AAAAGUGCUUACAGUGCAGGUAGACCUGCACUGUAAGCACUUUUUU; miR-106a-5p inhibitor (5’ to 3’): CUACCUGCACUGUAAGCACUUUU.
Statistical analysis
All representative data comes from experiments with similar results which were repeated at least three times and we used means ± standard deviation to present the collected data. Independent unpaired two-tailed Student’s t-tests were used to analyze the difference within the two groups and one-way ANOVA followed by Student-Newman-Keuls post hoc tests were used to detect differences in multiple groups. When p value < 0.05, the difference was considered to be statistically significant. * (p < 0.05) or ** (p < 0.01) were used to indicate the significances of difference within the two groups.
{kind=link}