Chemistry
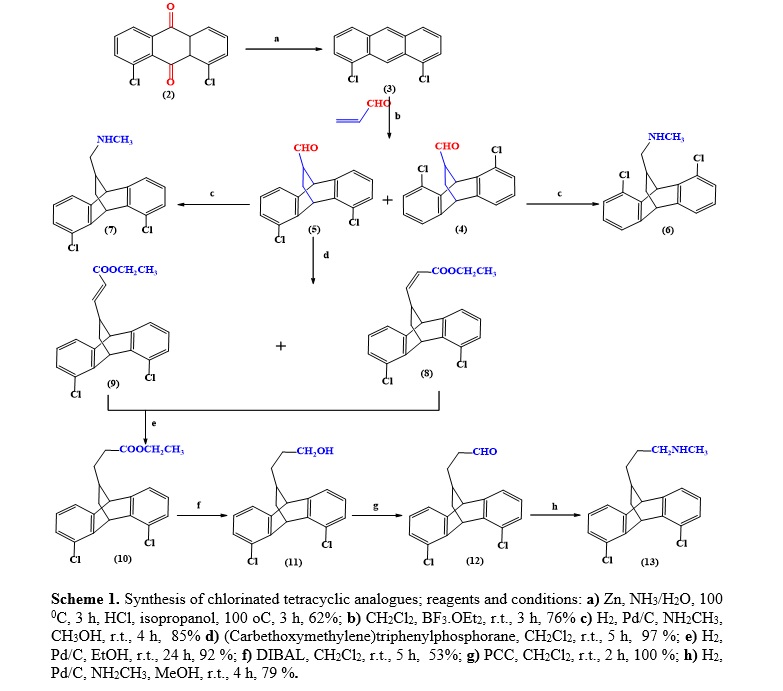
We recently reported a synthetic route to obtain the maprotiline analogues 1-(4,5-Dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)-N-methylmethanamine (6) and 1-(1,8-Dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)-N-methylmethanamine (7) (14, 15) and in our continuation interest in Diels-Alder reactions as well tetracyclic compounds (16-19) we developed a synthetic approach toward chlorinated tetracyclic maprotiline analogue 3-(1,8-Dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)-N-methylpropan-1-amine (13). Referring to the scheme 1; The reduction of the commercially available starting material 1,8-dichlroroanthraquinone (2) to afford 1,8-dichloroanthracene (3) was the first step toward the forward synthesis of the target compounds 6, 7 and 13. In 1973 House et al. reported that 1,8-dichloroanthracene 3 was prepared by reducing 1,8-dichloroanthraquinone 2 with zinc powder in aqueous ammonia followed by an acidic treatment (20). Later Zhao et al., reported that reduction could be accomplished with NaBH4 in isopropanol (21). In this work, 1,8-dichloroanthracene 3 was obtained in good yield according to House et al. method (20). The 1,8-dichloroanthracene 3 reacted with acrolein via Diels-Alder [4+2] cycloaddition reaction in dichloromethane at room temperature in the presence of boron trifluoride etherate as catalyst to afford a mixture of the intermediate isomers 4,5-dihalo-9,10-dihydo-9,10-ethanoanthracene-11-carbaldehyde (4) and 1,8-dihalo-9,10-dihydo-9,10-ethanoanthracene-11-carbaldehyde (5). The obtained isomers 4 and 5 is due to the fact that both the 1,8-dichloroanthracene (diene) 3 and acrolein (dienophile) being unsymmetrical. The mixture of carbaldehyde isomers was chromatographed on silica gel with the eluent system ethyl acetate: petroleum ether (1:10) and carbaldehyde 4 was eluted first. The maprotiline analogues 6 and 7 were obtained by direct reductive amination of their respective carbaldehydes 4 and 5 respectively. The reductive amination of carbaldehydes 4 and 5 was carried out separately by treating with 3 molar equivalents of a commercially available solution of methylamine in methanol in the presence of Pd-C as heterogeneous catalyst and stirred for 4 hours at room temperature under H2 (balloon). After filtration of the reaction mixture through a pad of celite and evaporation of the solvent, the corresponding amine analogues 6 and 7 were obtained. The carbaldehyde 5 was further recruited and converted by Wittig homologation using (carbethoxymethylene)triphenylphosphorane into two carbon homologated α,β unsaturated ester isomers namely; Z-Ethyl 3-(1,8-dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)propenoate 8 and E-Ethyl 3-(1,8-dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)propenoate 9. The Wittig reaction was smoothly carried out at room temperature for 5 hours in dichloromethane affording 97% of α,β-unsaturated ester isomers 8 and 9. These isomers were easily separated by silica gel column chromatography with the eluent system ethyl acetate: petroleum ether (1:10). The NMR J-coupling and chemical shift of the vinylic protons were employed to distinguish between the Z 8 and E 9 isomers. The ratio of the isomers, as deduced from integration of the vinylic proton signals, is approximately 1:2. The 1H-NMR spectrum of the Z 8 showed double doublet signal at δ 5.47 ppm with coupling constants J = 11.3, 9.5 Hz integrated for the proton assigned for olefinic proton (-CH=CH-) and a doublet signal at δ 5.61 ppm with coupling constant J = 11.7 Hz integrated for the proton assigned for olefinic proton (-CH=CH-) attached to ester group (-COO-CH2-CH3). Whereas these signals of the E 9 appeared at δ 6.36 ppm as double doublet with coupling constants J = 15.4, 9.5 Hz and at δ 5.75 ppm as doublet with coupling constant J = 15.4 Hz. The α,β unsaturated esters 8 and 9, products of the Wittig reaction, were then subjected to hydrogenation to reduce the double bond by stirring for 24 hours at room temperature in ethanol in presence of Pd/C under H2 (balloon). After filtration of the reaction mixture through a pad of celite and solvent was removed in vacuo, the saturated ester Ethyl 3-(1,8-dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)propanoate 10 was obtained in an excellent yield of 92 %. Reduction of ester 10 with reducing agent diisobutylaluminium hydride (DIBAL) at room temperature gave the alcohol 3-(1,8-Dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)propan-1-ol (11) in 53 % yield, which was completely oxidized using Pyridinium Chlorochromate (PCC) at room temperature in dichloromethane to give the desired aldehyde 3-(1,8-Dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)propanal (12). An attempt to transform the ester 10 to the aldehyde 12 in one step according to the known literature procedure (22) using DIBAL at -78 0C resulted in a mixture of aldehyde 12 and its corresponding alcohol 11. Direct reductive amination of the aldehyde 12 by the same procedure applied to synthesize the chlorinated maprotiline analogues 6 and 7 led to the desired chlorinated maprotiline analogue 13. The overall yield of the synthesis of the target (76) using DIBAL at – 78 0C and at room temperature was 19 % and 15.3 % respectively.
In vitro anticancer activity evaluation
The cancer cell lines were incubated with serial dilution of each compound (from 313 pg ML-1 to 5 mg ML-1) in a 96-well plate for 4 days, and then tested for growth inhibition by MTT-Test. Maprotiline 1 had been used as a positive control. Previous studies reported that maprotiline showed a potential antiproliferative activity against BL lymphoma cell line DG-75 (6, 7). A number of 9,10-dihydro-9,10-ethanoanthracenes were exhibited potent antiproliferative activity through inducing apoptosis and caspase activation in BL cell lines as well some of these compounds displayed activity in multi-drug resistant (MDR) cells. Furthermore many of these compounds were more active than maprotiline (8). Our results showed that all tested maprotiline analogues as well intermediates were able to inhibit the growths of the cancer cell lines A549 and HePG2 at low micromolar concentrations. In addition, the intermediates compounds 4 and 5 were also able to inhibit the growth of a third cell line (HCT). The IC50 values of all compounds, and that from maprotiline, are given in Table 1. The chlorinated maprotiline analogues 6, 7 and 13 were found to exhibit a potent antiproliferative effect on A549 cell line with IC50 values 25.5, 18.9 and 7.8 µg/ml respectively as well as against HePG2 cell line with IC50 values 12.66, 13.8 and 4.44 µg/ml while these maprotiline analogues had no effect on HCT cell line. The results showed that intermediates compounds 4 and 5 with formyl group were more potent than compounds 6, 7 and 13 against A549 and HepG2 cell lines, the IC50 of compound 4 and 5 were 1.1 and 3.71 against A549 and 0.12 and 0.65 µg/ml against HepG2 respectively. that’s mean the sensitivities of the treated cancer cells to 4 and 5 were 6 times higher than maprotiline in case of the breast cancer cell line A459, and even 40 times higher in case of hepatocyte carcinoma cell line HepG2. Furthermore, The IC50 of compounds 4 and 5 against HCT were 0.4 and 0.7 µg/ml respectively. Importantly the intermediates compound 4 with formyl group above on chlorine atom was the most potent against all three tested cancer cell lines. This result shows a direct or an indirect role of the formyl group and its position in the biological activity of the two compounds 4 and 5. Further investigations for these compounds are suggested.
Table 1. The IC50 of the Tested Compounds
|
Substance
|
A549
|
HepG2
|
HCT
|
|
IC50 value µg/ml
|
IC50 value µg/ml
|
IC50 value µg/ml
|
|
4
|
1.1( 0.2)±
|
0.12 (±0.03)
|
0.4 (±0.1)
|
|
5
|
3.71 (±0.8)
|
0.65 (±0.1)
|
0.7 (±0.1)
|
|
6
|
25.5 (±5.2)
|
12.66 (±4.4)
|
ND
|
|
7
|
18.9 (±2.5)
|
13.8 (±0.6)
|
ND
|
|
13
|
7.8 (±1.25)
|
4.44 (±0.41)
|
ND
|
|
Maprotiline
|
6.1 (±1.16)
|
5.15 (±0.77)
|
ND
|
ND=Not determined
Experimental
Synthetic procedures
4.1.1. Synthesis of :1,8-Dichloroanthracene (3), 4,5-Dichloro-9,10-dihydro-9,10-ethanoanthracene-11-carbaldehyde (4), 1,8-Dichloro-9,10-dihydro-9,10-ethanoanthracene-11-carbaldehyde (5), 1-(4,5-Dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)-N-methylmethanamine (6) and 1-(1,8-Dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)-N-methylmethanamine (7).
The compounds 3-7 were synthesized according to (14, 20) and characterization is also recorded in supplementary data.
4.1.2. Synthesis of Z-Ethyl 3-(1,8-dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)propenoate (8) and E-Ethyl 3-(1,8-dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)propenoate (9)
(Carbethoxymethylene)triphenylphosphorane (2 g, 5.75 mmol) was added to a solution of the aldehyde 5 (1.2 g, 4 mmol) in (36 ml) CH2Cl2. The reaction mixture was stirred at room temperature for 5 h. The solvent was removed and the residue was purified via flash column chromatography on silica gel using (Ethyl acetateA/Petroleum ether, 1:5) to afford separable isomers 8 and 9 (1.45 g, 97 %) in ratio of 1:2 respectively as yellow oil.
Compound 8: IR (KBr): ν = 3066, 2927, 2860, 1716, 1641, 1575, 1456, 1190, 1029, 771, 759,594 cm-1; 1H NMR (CDCl3, 400 MHz): δ = 1.20 (t; J = 7.3, 3H, -O-CH2-CH3), 1.24-1.29 (m; 1H, H-12) 2.11-2.14 (m; 1H, H-12), 3.76 (m; 1H, H-11), 4.08 (q; J = 7.3, 2H, -O-CH2-CH3), 4.18 (d; J = 2.9, 1H, H-10), 5.29 (t; J = 2.9, 1H, H-9), 5.47 (dd; J = 11.3, 9.5, 1H, -CH=CH-COO-), 5.61 (d; J = 11.7, 1H, -CH=CH-COO-), 6.96-7.19 (m; 6 H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ =14.2, 33.1, 36.5, 36.9, 49.7, 60.0, 119.6, 122.1, 123.7, 126.6, 126.8, 127.0, 129.3, 129.5, 139.5, 140.3, 142.3, 145.2, 152.2, 166.0 ppm; MS (EI): m/z (%) = 372 (10) [M+], 367 (5), 248 (65), 246 (100), 176 (18), 131 (5), 69 (12); HRMS (EI): Calcd. For C21H18O2Cl2 [M+] 372.0684, Found 372.0683.
Compound 9: IR (KBr): ν = 3066, 2979, 2935, 2898, 1718, 1650, 1577, 1456, 1446, 1369, 1271, 1180, 1039, 985, 769, 740, 703, 590 cm-1; 1H NMR (CDCl3, 400 MHz): δ = 1.22 (t; J = 7.3, 3H, -O-CH2-CH3), 1.22-1.24 (m; 1H, H-12) 2.04-2.07 (m; 1H, H-12), 2.71 (m;1H, H-11), 4.12 (q; J = 7.3, 2H, -O-CH2-CH3), 4.20 (d; J = 2.2, 1H, H-10), 5.35 (t; J = 2.5, 1H, H-9), 5.75 (d; J = 15.4, 1H, -CH=CH-COO-), 6.36 (dd; J = 15.4, 9.5, 1H, -CH=CH-COO-), 7.04-7.24 (m; 6 H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 14.2, 32.0, 36.8, 40.9, 49.7, 60.3, 121.5, 121.9, 123.8, 126.7, 126.9, 127.0, 127.1, 129.3, 129.8, 139.5, 140.2, 141.6, 145.1, 150.5, 166.2 ppm; MS (EI): m/z (%) = 372 (41) [M+], 367 (19), 248 (62), 246 (100), 176 (29); HRMS (EI): Calcd. For C21H18O2Cl2 [M+] 372.0684, Found 372.0683.
4.1.3. Synthesis of Ethyl 3-(1,8-dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)propanoate (10)
In a two-necked round-bottomed flask (0.37 g of 10% Pd/C) was wetted with ethanol and the flask was evacuated, and flushed with hydrogen two times, then a solution of (1.3 g, 3.5 mmol) unsaturated ester 8 and 9 in (40 ml) ethanol was added to the reaction mixture. The mixture was stirred for 24 h at room temperature under H2 (balloon). The reaction mixture was filtered through a pad of celite and the solvent was removed in vacuo to afford the corresponding 10 (1.2 g, 92%) as yellow oil.
IR (KBr): ν = 3020, 2933, 2900, 1733, 1460, 1375, 1261, 1176, 1029, 754, 559 cm-1; 1HNMR (CDCl3, 400 MHz): δ = 1.12-1.16 (m; 2H, H-/1), 1.24 (t; J = 7.3, 3H, -O-CH2-CH3), 1.46-1.51 (m; 1H, H-12), 1.86-1.93 (m; 1H, H-11), 1.97-2.04 (m; 1H, H-12), 2.31 (t; J = 8.0, 2H, H-/2), 4.08 (q; J =7.3, 2H, -O-CH2-CH3), 4.15 (d; J = 2.2, 1H, H-10), 5.29 (t; J = 2.5, 1H, H-9), 6.99-7.25 (m; 6 H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ =14.1, 31.1, 32.4, 34.3, 37.9, 44.1, 48.7, 60.2, 122.9, 123.2, 123.3, 125.2, 125.4, 125.5, 125.5, 125.8, 140.4, 143.2, 143.7, 144.2, 173.4 ppm; MS (EI): m/z (%) = 374 ([M+], not recorded), 331 (11), 329 (25), 295 (12), 248 (58), 246 (100), 212 (46), 178 (45).
4.1.4. Synthesis of 3-(1,8-Dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)propan-1-ol (11)
To a solution of saturated ester 10 (600 mg, 1.6 mmol) in CH2Cl2 (6 ml), DIBAL (7 ml) was added. The reaction mixture was stirred for 5 h at room temperature. Then the reaction mixture was quenched with Methanol (1 ml) followed by the addition of ethyl acetate (30 ml) and saturated aqueous of NH4Cl (10 ml). The quenched reaction mixture was filtered through suction funnel and extracted with CH2Cl2 and the combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified via flash column chromatography on silica gel using ethyl acetate / hexane (1:3) to afford 11 (280 mg, 53 %) as a milky viscous oil.
IR (KBr): ν = 3577, 3336, 2970, 2933, 2860, 1456, 1055, 756, 567cm-1; 1HNMR (CDCl3, 400 MHz): δ = 0.87-0.98 (m; 2H, H-/1), 1.13-1.28 (m; 1H, H-12), 1.53-1.63 (m; 1H, H-11), 1.84-1.93 (m; 2H, H-/2), 1.97-2.08 (m; 1H, H-12), 3.47 (t; J = 6.6, 2H, -CH2OH), 4.04 (d; J = 2.2, 1H, H-10), 4.16 (t; J = 2.5, 1H, H-9), 7.01-7.17 (m; 6 H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 30.6, 32.2, 34.6, 38.2, 44.3, 48.9, 62.8, 122.9, 123.1, 123.3, 125.2, 125.3, 125.4, 125.5, 125.6, 140.7, 143.3, 143.8, 144.4 ppm; MS (EI): m/z (%) = 332 ([M+], not recorded), 295 (7), 264 (12), 212 (29), 178 (100), 1152 (4).
4.1.5. Synthesis of 3-(1,8-Dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)propanal (12)
To a solution of alcohol 11 (250 mg, 0.75 mmol) in CH2Cl2 (6 ml), PCC (250 mg, 1.2 mmol) was added. The reaction mixture was stirred for 2 h at room temperature. The reaction mixture was concentrated in vacuo and the residue was purified via flash column chromatography on silica gel using ethyl acetate / hexane (1:3) to afford 12 (250 mg, 100 %) as a colorless oil.
IR (KBr): ν = 3020, 2935, 2862, 2812, 1726, 1460, 1172, 1026, 760, 754, 559 cm-1; 1HNMR (CDCl3, 400 MHz): δ = 1.12-1.25 (m; 2H, H-/1), 1.43-1.50 (m; 1H, H-12), 1.84-1.90 (m; 1H, H-11), 1.97-2.04 (m; 1H, H-12), 2.41-2.45 (m, 2H, H-/2), 4.10 (d; J = 2.2, 1H, H-10), 4.25 (t; J = 2.9, 1H, H-9), 7.09-7.25 (m; 6 H, ArH), 9.69 (t; J = 1.4, 1H, CHO) ppm; 13C NMR (CDCl3, 100 MHz): δ = 28.3, 34.5, 37.9, 41.9, 44.1, 48.7, 123.0, 123.2, 123.4, 125.6, 125.5, 125.6, 125.6, 125.9, 140.2, 143.1, 143.7, 144.0, 202.1 ppm; MS (EI): m/z (%) = 330 ([M+], not recorded), 321 (7), 319 (11), 311 (12), 289 (7), 251 (6), 225 (8), 204 (11), 201 (21), 199 (51), 197 (92), 181 (28), 165 (100), 151 (38), 149 (15).
4.1.6. Synthesis of 3-(1,8-Dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)-N-methylpropan-1-amine (13)
In a two-necked round-bottom flask (100 mg, 10% Pd/C) was wetted with dichloromethane and the flask was evacuated, flushed with hydrogen two times, then a solution of (110 mg, 0.33 mmol) aldehyde 12 in (5 ml) methanol was added to the reaction mixture followed by the addition of (0.7 ml, 2 M) solution of methylamine in methanol. The mixture was stirred for 4 h at room temperature under H2 (balloon). The reaction mixture was filtered through a pad of celite and the solvent was removed in vacuo to yield (90 mg, 79 %) of the corresponding amine 13 as white powder, mp: 178 0C.
IR (KBr): ν = 3414, 2935, 2864, 1471, 1399, 1171, 1034, 804, 752, 551, 466 cm-1; 1HNMR (CDCl3, 400 MHz): δ = 0.79-0.87 (m; 2H, H-/1), 1.0-1.10 (m; 2H, H-/2), 1.2 (s; 3H, N-CH3), 1.73-1.79 (m; 1H, H-11), 1.85-1.92 (m; 2H, H-12), 2.68-2.72 (m; 2H, H-/3), 3.18 (d; J = 1.8, 3H, N-CH3), 4.03 (d; J = 2.2, 1H, H-10), 4.13 (t; J = 2.2, 1H, H-9), 6.97-7.02 (m; 3H, ArH), 7.13-7.18 (m; 3H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 23.8, 32.8, 32.9, 34.4, 37.7, 44.1, 48.5, 49.2, 122.9, 123.3(2x), 125.2, 125.4, 125.5, 125.6, 125.8, 140.3, 143.2, 143.6, 144.0 ppm; MS (EI): m/z (%) = 346 (18) [M+ + H], 336 (19), 335 (31), 334 (75), 332 (100), 326 (5), 318 (11), 298 (11), 286 (5), 284 (9); HRMS (EI): Calcd. For C20H22NCl2 [M+] 346.1129, Found 346.1128.
MTT assay protocol
In vitro anti-cancer activity was demonstrated by determining the IC50 values in three cancer cell lines, including the lung carcinoma cell line A549, the hepatocellular carcinoma HepG2 cell line, and the colorectal carcinoma HTC-116 cell line (Table 2.). Growth inhibitions were measured in 96-well plates. Aliquots of 120 µL of the suspended cells (50,000 mL-1) were given to 60 µL of a serial dilution of the inhibitor(S). After 5 days of incubations, growths were determined the MTT assay (MTT= 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide). Briefly, 20 μl MTT (5 mg/mL in PBS) were added to each well, and the plates were incubated for 2 h at 37ºC, and 5% CO2-atmosphere in the cell incubator. The supernatants were then discarded and 200 μl of isopropanol/ HCl were added to each well and mixed to dissolve the formazan crystals. The absorbance was then read at 550 nm using a microplate reader (Thermo Scientific, USA). The viability of the cells was calculated by dividing the absorbance average of the treated cells by the absorbance average of the control cells multiply 100%. The IC50 values were defined as sample concentration inhibiting 50% of cell growth. The activities of the cells were plotted against the concentration of the drugs, and the IC50 values were calculated from the regression curve.
Table 2. Cancer Cell Lines
|
No.
|
Cell line
|
ATCC-No.
|
Disease or organ
|
|
1
|
A549
|
A549 (ATCC® CCL-185™)
|
Lung Carcinoma
|
|
2
|
HepG2
|
HepG2 [HEPG2] (ATCC® HB-8065™)
|
Hepatocellular carcinoma
|
|
3
|
HTC-116
|
HCT 116 (ATCC® CCL-247™)
|
Colorectal Carcinoma
|
{kind=link}