Patients and samples collection
The study protocol was approved by the Human Ethics Committee of the Fudan University Obstetrics and Gynecology Hospital, and all participants provided written informed consent. The patients included in this study were early pregnant women (age 20-38 years; gestational age 7-9 weeks) who voluntarily requested termination of pregnancy in Fudan University Obstetrics and Gynecology Hospital from May 2018 to August 2019. Normal decidual tissue specimens (n = 40), normal villi tissue (n = 14), and villi of RSA patients (n = 10 (Fig. 4d)) were collected. Normal pregnancy patients had no history of spontaneous abortion, stillbirth and other adverse pregnancies. Ultrasound showed normal embryo development and primitive fetal heart beat (+). There were no symptoms and signs of threatened abortion such as vaginal bleeding and abdominal pain during this pregnancy. RSA patients had two or more consecutive spontaneous abortions before 24 weeks of gestation. The decidual tissues were stored in ice-cold DMEM / F-12 (HyClone, USA) under sterile conditions and transported to the lab within 1 hour after surgery. Then, the DIC primary cells were isolated and cultured. RSA patients' villi and normal villi were immersed in electron microscopic fluid for subsequent testing.
dNK cells isolation and cell culture
The decidual tissues were washed in PBS (HyClone), then cut into 1 mm3 pieces, digested with 20% type IV collagenase (0.1%; Sigma-Aldrich, USA) and 5% DNA enzyme (3000IU, Sigma, Germany) at 37 ° C for 30 minutes. The tissue fragments were filtered through sieves (pore size: 100, 300 and 400 mesh), centrifuged at 1300 rpm for 10 minutes, and the supernatant was discarded. 20%, 40%, and 60% Percoll (Amersham, USA) were prepared, and then the lower layer DIC was recovered by density gradient centrifugation at 2500 rpm for 30 minutes. DIC was cultured overnight in RPMI-1640 medium (HyClone) containing 10% fetal bovine serum (FBS, Gibco, USA). According to the manufacturer's instructions, NK cells were negatively selected from DIC using human NK cell separation kits (MACS, Miltenyi Biotec, Germany). The dNK purity measured by FCM was over 90%.
NK cells were cultured in 1640 (HyClone, USA) containing 10% FBS (Gibco) and 1% penicillin-streptomycin solution (HyClone, USA). Furthermore,IL-2 (20ng/μl), IL-15 (20ng/μl) and IGF-2 (50ng/μl) were supplemented according to different experiments. HTR-8/SVneo cell lines were cultured in DMEM/F12 (HyClone) containing 10% FBS (Gibco) and 1% penicillin-streptomycin solution (HyClone).
Co-culture of dNK cells and HTR-8/SVneo
HTR-8 / SVneo was pretreated with or without rapamycin (2μM, Sigma, USA) for 48 hours in a 24-well plate (Corning, USA) and then co-cultured with dNK cells. In addition, HTR-8 / SVneo was pretreated with or without 3-MA (10mM, Sigma, USA) for 24 hours and then co-cultured with dNK cells. The ratio of dNK cells (2 × 105 cells/well) to HTR-8 / SVneo (1 × 105 cells/well) was 2:1. After 48 hours of co-culture, all suspended cells in the co-culture system were collected for subsequent experiments.
Transmission electron microscopy
The villi of RSA patients and normal abortion patients were collected, and the tissues' volume were generally not more than 1mm3. Fresh tissues were quickly placed into the electron microscope fixative (Servicebio), fixed at 4 °C for 2-4 h, and post-fixed in 1% osmium acid for 2 h. The samples were dehydrated in a series of gradient concentration alcohols, permeated overnight with a mixture of acetone and 812 embedding agent (SPI) (2:1), then embedded in pure 812 embedding agent and polymerized in a 60 °C oven for 48 h. Samples were cut into 60-80 nm ultrathin sections by using ultrathin slicing machine (Leica UC7). The sections were double stained with uranium and lead (2% uranyl acetate saturated alcohol solution, lead citrate) and dried overnight at room temperature. Finally, observed under a transmission electron microscope (HITACHI, HT7700), collected images for analysis.
Lentiviral infection
HTR-8 / SVneo cells were seeded in a six-well plate (5 × 104 cells/ml). After confluence reached 30%, the cells were infected with ATG5 silencing lentivirus (ATG5-RNAi)/PEG10 overexpression lentivirus (PEG10over), and their corresponding negative control virus (NC) (all from Genechem Co., LTD.). According to the manufacturer's instructions. (see Equation 1 in the Supplementary Files)
The optimal infection condition of MOI was 80% of infected cells in the best time. Calculated the required virus volume using the formula, after 12 hours of the cells were infected, the medium was changed and continued to culture. The infection efficiency was observed under a fluorescence microscope at 48 h and 72 h after infection, and subsequently screened for 1 week using 1 μg/ml puromycin (Genechem Co., LTD.).
qRT‐PCR
The cells or mouse placental tissues were collected, and total RNA was extracted by RNAiso Plus reagent (TaKaRa Biotechnology). According to the manufacturer's instructions, 1000 ng of total RNA was reverse transcribed into cDNA with a reverse transcription kit (TaKaRa Biotechnology). Subsequently, detection was carried out on a real-time PCR instrument (ABI QuantStudio 6 Flex, USA). Reaction system (10μl): 5μl TB Green Premix Ex TaqTM II, 0.2μl ROX Reference Dye II, 1μl cDNA, 0.4μl Forward Primer, 0.4μl Reverse Primer and 3μl RNase Free dH2O. Primers were listed in Table 1. Reaction conditions (40 cycles): denaturation (95 °C 30 s), annealing (95 °C5 s) and elongation (60 °C 34 s). Finally, the infection efficiency of lentivirus siATG5, PEG10over and the expression of related molecules were analyzed using the 2-ΔΔCT method.
Table 1. Related primer sequences
Transcriptome sequencing
HTR-8/SVneo cells transfected with ATG5-RNAi (n=3) and NC (n=3) were added to RNAiso Plus reagent (TaKaRa), hereafter submitted to Shanghai Litzchi Biosystems (LITCHI BIO, Shanghai, China) for subsequent RNA-seq analysis. Specific processes include RNA extraction, RNA sample quality inspection, library construction, library purification, library detection, library quantification, sequencing cluster generation, and sequencing on the Hiseq 4000 platform. FastQC software (V0.10.1) was used to control the quality of the offboard data, and then DESeq2 (V1.6.3) of Bioconductor software package was used to analyze and screen the differential genes. The differentially expressed gene standards were as follows: expression amount fold difference threshold |logFC|>1; expression difference significance threshold P-value < 0.05. The results of sequencing were analyzed and produced a related thermograms using pheatmap Version 1.0.8 in R3.4.1. In addition, based on the STRING database, the protein interaction relationship between the differential genes and the interest genes (NK function-related genes, invasion-related genes, autophagy-related genes) were predicted, and then the network maps were constructed via the Cytoscape software.
Protein extraction and western blotting
After the cells were washed with PBS, lysate (RIPA: 100 XPMSF = 100:1) (Beyotime, china) was added, and the cells were separated by cell scraper after 30 minutes on ice, centrifuged at 12,000 rpm for 30 min at 4 °C. Then the supernatant was collected and the protein concentration was determined by BCA Protein Assay Kit (Beyotime, China). 1 / 4 of 5XSDS-PAGE (Beyotime, china) was added according to the protein volume, boiled at 99 ° C for 10 minutes, and then store at -80 ° C. In 12.5% SDS-PAGE (Epizyme, Shanghai, China), the total protein (30 µg / pore) was electrophoretic and transferred to the PVDF membrane (Millipore, USA). The membrane was sealed at room temperature for 2 hours with 5% skim milk, and then washed 3 times for 15 minutes each using TBST (Sangon Biotech). Incubated with the primary antibodies against P62 (1:1000; Cell Signaling Technology,USA), LC3B (1:1000; Cell Signaling Technology, USA), ATG5 (1:3000;Abcam, Cambridge, UK), PEG10(1:5000;Abcam, Cambridge, UK), GAPDH (1:1000; Cell Signaling Technology,USA), α-Tubulin (1:2000;Proteintech,USA) overnight at 4 °C. Thereafter, the membranes were washed again 3 times with TBST, then incubated with horseradish peroxidase (HRP) conjugated goat-anti-rabbit IgG secondary antibody (1:3000; Cell Signaling Technology, USA) for 1 hour at room temperature. Finally, the membrane was washed 3 times and subjected to chemiluminescence treatment using an ECL Detection Kit (Millipore, USA).
Flow cytometry (FCM)
dNK cells co-cultured with HTR-8 / SVneo were collected from 24-well plates and centrifuged at 1300 rpm for 8 min. According to the recommended dose, dNK cells were stained with fluorescent dye-conjugated antibody of human antigen at 4 °C for 30min, including APC-conjugated anti-human CD56, FITC-conjugated anti-human CD16, APC/CY7-conjugated anti-human CD107a, PE-conjugated anti-human NKG2D/ Granzyme B, PE/CY7 -conjugated anti-human NKP30, and BV421-conjugated anti-human NKP46/IFN-γ (all from BioLegend), or the isotype control. Among them, the intracellular molecules (Granzyme B, IFN-γ) were added to FOXP3 Fix/Perm Buffer (4X) (BioLegend) to fix and break the membrane, then stained with antibody for 30min. After that, the cells were washed twice with PBS and resuspended. The samples were tested using a CyAN ADP analyzer (Beckman Coulter, USA) and analyzed by FlowJo software (TreeStar, USA). In isotype matched controls, the statistically labeled positive cells should be less than 5%.
Mice NK cells were used PE-conjugated anti-mouse NK1.1, APC/Cy7-conjugated anti-mouse CD3, PerCP/Cy5.5-conjugated anti-mouse CD16, FITC-conjugated anti-mouse NKG2D, APC-conjugated anti-mouse NKP46/IFN-γ, BV421-conjugated anti-mouse CD107a and FITC-conjugated anti-human/mouse Granzyme B (all from BioLegend) to stain. The samples were then analyzed by flow cytometry.
Immunohistochemistry
The paraffin sections of Human villi (5 μ m) were dehydrated in graded ethanol, next the endogenous peroxidase was removed with 3% hydrogen peroxide and incubated with 5% BSA at room temperature for 1h. After that, the samples were incubated with rabbit anti--human IGF-2 (1:200; abcam); rabbit anti--human PEG10 (1:500; abcam) or rabbit IgG isotypes at 4 °C overnight. After washing with PBS for three times, the sections were incubated with HRP-labeled secondary antibody at room temperature, reacted with 3,3-diaminobiphenylamine (DAB), and finally counterstained with hematoxylin.
Cell invasion assay
Matrigel (BD Bioscience) was diluted at a ratio of 1:8, and 35μL was added to the transwell upper chamber (8μm, Corning). The transwell chambers were placed in a 24-well plate for overnight stay at 4 °C. 200μL (HTR-8 / SVneo, 2 × 104 cells/well) DMEM / F12 suspension without FBS was added to the upper chamber, and 600μL DMEM / F12 containing 10% FBS was added to the lower chamber. According to different experimental requirements, 3-MA was added or not in the upper chamber, dNK cells (1×105 cells/well) were added or not in the lower chamber. The cells were cultured for 48h at 37 °C, 5% carbon dioxide incubator. The 24-well plate was removed, and the upper chamber medium and non-penetrating cells were gently wiped off with a cotton swab, PBS washed 3 times, fixed with 4% paraformaldehyde for 30 min, and crystal violet stained for 20 min. Thereafter, random photographs were taken under an inverted microscope (×100), and each chamber counted 5 visual fields. The number of invaded cells was counted by ImageJ software.
The cell-counting kit-8 (CCK-8) assay
CCK-8 assay (Dojindo, Tokyo, Japan) was used to detect the proliferation ability of HTR-8/SVneo cells after co-culture. The cells were seeded in 96-well plates (5 x 103 cells/well) for 0h, 24h, 48h and 72h, respectively. Then, 10μl CCK-8 solution was added to each well and cultured for 2 hours, the absorbance value at 450 nm was measured by microplate reader. Six parallel holes were set for each experiment and repeated three times.
In vivo experiments (Mice)
All mice used in the experiment were C57BL/6J strain (Shanghai jiesijie experimental animal Co., Ltd.) and were raised in an SPF experimental animal facility. 8-week-old female mice and 8-week-old male mice were caged at 2:1, and the pregnancy was confirmed on the day when the vaginal plug was seen (0.5 days). Pregnant mice were randomly divided into control group and experimental group. The experimental group was intraperitoneally injected with 3-MA (100 mg / kg / time, Sigma) at day 0.5, day 4.5, day 10.5, and the control group was injected with saline of equal volume at the same days. At day 8.5, the same amounts of pregnant mice in the control group and the experimental group were killed, and the number of embryo implantation and embryo absorption in the two groups were recorded respectively. The decidua cells of mice uterus were obtained by shredding, digesting, filtering and centrifuging. The relative molecular expression of NK cell was analyzed by FCM after anti-mouse immunofluorescent antibody staining. Similarly, the mice in the control group and the experimental group were randomly killed at day 14.5. In addition to recording the number of embryo implantation and embryo absorption, the endometrium of the embryo also needed to be peeled off, the development of the embryo was observed, and the crown-rump length of embryo and the weight of placenta were recorded respectively.
Statistical analyses
The results of at least three independent experiments were analyzed using the Graphpad Prism 6 (GraphPad, CA, USA) statistical software package. When the data was normally distributed, the two groups were analyzed by paired or unpaired t-test, and one-way ANOV was conducted between multiple groups. When the data was non-normally distributed, the Mann-Whitney rank sum test, the Wilcoxon paired test or the Kruskal–Wallis test were generally used. Data were expressed as mean ± SEM, and considered statistically significant when P < 0.05.
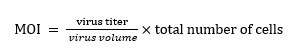{kind=link}