Identification of genomic regions specifically methylated in ovarian cancers
To identify genomic regions specifically methylated in ovarian cancers, from the 408,263 probes evaluable with adequate signals, we first selected 125,630 unmethylated probes (β value < 0.2) in two normal fallopian tube epithelial samples and two female leukocyte samples (Fig. 1). Then, from these 125,630 probes, we selected 15 probes in eight genomic regions methylated in 11 or more of 14 high-grade serous ovarian cancer cell lines (β > 0.8) and 21 or more of 30 ovarian cancer clinical samples (β > 0.2). From the eight genomic regions, we selected five genomic regions from four genes that had multiple (≥ 2) flanking probes which met the above criteria.
In addition, we evaluated copy number alterations of the four genes in ovarian cancers from a previous report [3] because such alterations could affect methylation levels and estimation of cancer cell fractions. Copy number alterations of the four genes were confirmed to be infrequent (< 2%). As a result, the four genes, PALLD, SIM1, OTX2, and ZNF154, were considered to be good candidate markers for ovarian cancer cell fractions (Table 1). Target CpG sites were located at a CpG island (CGI) or an N shore of a CGI (Fig. 2).
Methylation levels of the candidate marker genes in clinical samples
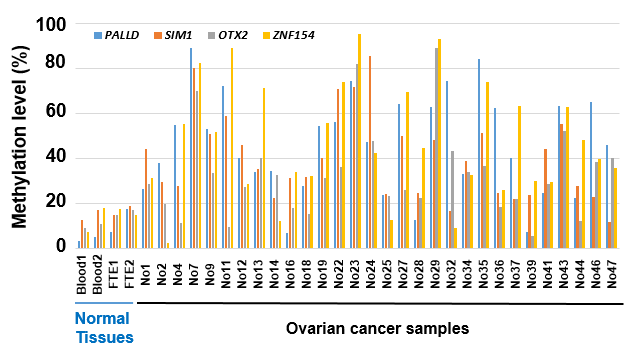
We next analyzed methylation levels of the four candidate marker genes in 30 clinical samples with information on their pathological cancer cell fractions, to evaluate whether these genes could reflect ovarian cancer cell fractions (Supplementary Fig. S1). The methylation level of OTX2 tended to be lower than those of the other three candidate marker genes, suggesting that OTX2 was not methylated in all of the ovarian cancer cells, and we excluded this gene from further analysis. For the remaining three genes, a correlation between the pathological cancer cell fraction and methylation level of a candidate marker gene was analyzed (Fig. 3). Correlation coefficients of PALLD, SIM1, and ZNF154 were 0.56, 0.61, and 0.71, respectively.
Implementation of pyrosequencing and confirmation of correlation
To estimate methylation levels more efficiently and cost-effectively, we tried to establish primers for pyrosequencing. However, we could not establish a primer set for PALLD because CpG sites in the region were too dense to establish primers with adequate specificity and sensitivity. For SIM1 and ZNF154, we were able to establish primers that linearly amplified partially methylated DNA samples with high correlations (R = 0.97 and P = 0.0004 for SIM1, and R = 0.98 and P = 0.0002 for ZNF154) (Fig. 4).
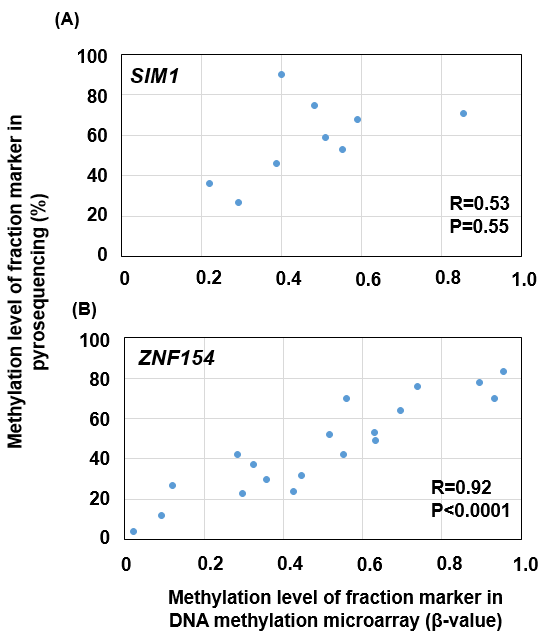
Using the primers, we performed pyrosequencing of the screening cohort samples with the remaining samples. Correlations between the b values in DNA methylation microarray and methylation levels in pyrosequencing were moderate for SIM1 (R = 0.53 and P = 0.55) and high for ZNF154 (R = 0.92 and P < 0.0001) (Supplementary Fig. S2). Therefore, we considered that, for ZNF154, methylation levels of the CpG sites analyzed by the pyrosequencing had a high correlation with b values of the CpG site analyzed by a DNA methylation microarray. In contrast, for SIM1, methylation levels of the CpG sites analyzed by the pyrosequencing had only a moderate correlation with b values of the CpG site analyzed by DNA methylation microarray.
Validation of the cancer cell fraction maker in an independent sample set
To validate whether ZNF154 methylation levels by pyrosequencing and pathological cancer cell fraction coincide, we performed pyrosequencing of 20 independent additional samples. Correlation between pathological cancer cell fraction that had already been determined by pathology of histological sections and the ZNF154 methylation level was 0.81 (P < 0.0001) (Fig. 5). This successful validation in an independent sample set showed that ZNF154 methylation level is as a good marker of ovarian cancer cell fraction as that determined from histological sections.
{kind=link}
{kind=link}