3.1 Molecular geometry
These anthraquinone compounds were subjected to geometry optimization in the ground state. The optimized molecular structure of the anthraquinone compounds with atom numbering scheme adopted in the present study is presented in Fig. 1. On compared with the optimized phase, geometric conformations of these compounds changed obviously after interacting with the DNA gyrase B at the active site. There are obvious changes about the bond angles and dihedral angles rather than the bond lengths, which suggested strong inter-molecular interactions. The corresponding structural parameters (bond length and dihedral angle) are listed in Supplementary Table S1. Compared with the optimized structure, it can be seen from the Table S1 that there is almost no change in the length of each bond of compounds at the active site. The glucose ring that binds to the aromatic ring ketone is obviously bent at the active site, ascribing to the more serious intermolecular interaction with surrounding amino acids (Wang et al., 2010). In general, almost all of the anthraquinone skeleton planes are stable, whereas the angles between the glucose groups and the phenyl ring are more or less changed (Fig. 1). This can be concluded that the interactions of these anthraquinone compounds might be related with the type and number of substituent groups on the molecular structure, as well as the substituted glucose rings.
3.2 Molecular docking
According to the literature (Devipriya and Kumaradhas, 2013; Zhu et al., 2021), the binding modes and binding affinities play important roles in the inhibition of the small molecules to the enzyme. Results indicated that the selected binding affinities for these complexes of molecules-DNA gyrase B are from -7.27 to -10.24 Kcal/mol. It is worth noting that the bonding energies of the anthraquinones are -8.50, -8.24, -7.27, -7.91 and -7.66 kcal/mol for molecules M1-M5, respectively. However, the corresponding anthraquinone glucoside compounds have generally higher intermolecular binding energy due to the strong interaction between the glucose ring and the surrounding amino acids Asn46, Arg76, Pro79, Gly101, and Lys103 (-10.24,-9.47,-9.74,-9.31 and -9.11 kcal/mol for molecules M6-M10, respectively). This is consisted with the experience result that the anthraquinone glucosides have much more antibacterial effect than their corresponding aglycones (Wang et al., 2010). At the same time, the structure of the glucose ring has also undergone significant changes (Table S1). Among them, the O1 of the glucose ring of the lowest binding energy molecule M6 (-10.24Kcal/mol) forms a hydrogen bond with the hydrogen atom HD21 of ASN46 at a length of 2.197Å, and the side hydroxyl group of the glucose ring forms hydrogen bond interactions with LYS103 and ASN46 at length of 2.091 Å and 2.062 Å, respectively (Fig. 2f).
Compared with the compound M6, the glucose ring at the active site of M7 rotates clearly clockwise, resulting in a smaller angle with the anthraquinone ring plane. The dihedral angle of C11-C12-O18-C26 is changed from 16.23º at the optimized state to -116.95º, which can be ascribed to the strong hydrogen bonding and electrostatic interactions with surrounding amino acids Asn46, Ala100, Phe104, Gly117 and Val120 (Fig. 2g). Besides the multiple hydroxyl groups on the anthraquinone skeleton, hydroxyl groups of the glucose ring are also important to the antimicrobial effect, which may be ascribed as the hydrogen bonding interactions at the active site. In fact, the combined anthraquinone chrysophanol 8-O-beta-D-glucoside shows good bioactivity on compared with the free chrysophanol (Li et al., 2007). Therefore, the influence of substituent groups on anti- DNA gyrase B activity might not only be related with the type and number of substituent carboxyl, hydroxyl and hydroxylmethyl on phenyl ring groups, but also the combined glucose rings. Compared with the hydroxyanthraquinones, the interact area of the whole anthraquinone glucosides are larger (the part shown by the purple circle in Fig. 2k), between which the hydrophobic interaction plays a very important role. The nearest neighbors and short contact distances (Å) of anthraquinone compounds with the amino acid residues of DNA gyrase B are shown in Table 1.
3.3 Molecular electrostatic potential
Molecular electrostatic potential (MEP) on molecular vdW surface is an important parameter to study the intermolecular interaction (Murray and Politzer, 1998; Murray and Politzer, 2011; Lu and Chen, 2012). Usually, MEP is also related to the electronic density (Vijayalakshmi and Suresh, 2008), thus it is always used to anticipate reactive sites that electrophilic and nucleophilic reagents attack (Selvaraju et al., 2012). In this text, the negative (green) and positive (red) regions are regarded as electrophilic and nucleophilic reactivity regions, respectively (Fig. 5), while the white regions shows zero potential. In general, the electrostatic potential distribution of anthraquinone compounds is more uniform than that of the anthraquinone glucosides. The main positive charge is distributed near the hydrogen atom of the hydroxyl group on the side chain of the aromatic ring plane. For the molecule M3, the oxygen atom O18 in the carboxyl group acts as an electron donor to form a hydrogen bonding interaction with the HH11 of the amino acid Arg136, and thence the region at vicinity of the connected hydrogen atom H28 remains the highest electrostatic potential value of 66.73Kcal/mol. Due to the strong interaction between the side chain hydroxyl oxygen atoms (O17 and O19) of molecule M1 and the surrounding amino acid Gly77, and the electrostatic interaction between the oxygen atom O15 of the anthraquinone ring and amino acid Arg76, the electrostatic potential has reached the lower value of -58.25 Kcal/mol (Fig. 3a).
Once glucose ring is introduced into the hydroxyanthraquinone plane, it was shown that almost all of the positive molecular potentials of anthraquinone glucosides are distributed among the hydroxyl hydrogen atoms of the glucose structures except the molecule M9. These substituent hydroxyl groups on the molecular aromatic ring interact with the amino acids Glu42, Asn46 and Gly11 by hydrogen bonding and electrostatic interaction, which might lead to imbalance of distribution of electrons (the value of the global surface maximum is 75.40 Kcal/mol, Fig. 3i). In general, due to the presence of the hydroxyl group in the glucose ring, it can interact with surrounding amino acids Lys103, Asn46, Ala100, Phe104 and Val120 by intermolecular hydrogen bonds, electrostatic attraction and Van der Waals forces, resulting in major changes in the molecular structure and the molecular electrostatic potential distribution. In general, the positive molecular potentials at the vicinity of the molecular hydrogen atom are increased obviously when glucose ring is introduced into the hydroxyanthraquinone ring. For example, the global surface maximum of M1 is 61.35 Kcal/mol while the value changed to be 70.18 Kcal/mol for M6, the value of M2 (44.37 Kcal/mol) increased about 8.19 Kcal/mol (52.56 Kcal/mol) for M8. This indicated that the electronic cloud of the aromatic side chain has shifted while interacting with the bonding site.
3.4 The molecular orbitals
The important Frontier Molecular Orbital (FMOs) are highest (HOMO) occupied molecular orbitals and the lowest (LUMO) unoccupied one, which play an important part in the chemical stability of the molecule (Lim et al., 2011). The HOMO represents the ability to donate an electron, while the electron acceptor LUMO represents the ability to accept an electron. The energy gap between HOMO and LUMO also determines the chemical reactivity, optical polarizability and chemical hardness-softness of the molecule (Huang et al., 2017). According to the Koopman's theorem, EHOMO and ELUMO values of any chemical type are associated with its ionization energy and electron affinity values, respectively (Plakhutin and Davidson, 2009). In addition, for closed-shell compounds, hardness (ɳ), chemical potential (μ) and electronegativity (χ) and softness are also defined. Recently, a new descriptor electrophilicity index (ω) has also been defined to quantity the global electrophilic power of the compound (Tandon et al., 2019). Dipole moment D (debye) is also shown in Table 2. The usefulness of this new reactivity quantity has been recently demonstrated in understanding the toxicity of various pollutants in terms of their reactivity and site selectivity (Abraham et al., 2019).
ɳ = (I-A)/2 (1)
μ= -(I+A)/2 (2)
χ = (I+A)/2 (3)
ω =μ2/2ɳ (4)
Where, A and I are the ionization potential and electron affinity of the compounds respectively.
Commonly, the atom occupied by more densities of HOMO should have stronger ability to detach an electron whereas the atom with more occupation of LUMO should have ability to gain an electron. In the present study, the HOMO and LUMO energies are predicted at B3LYP method with 6-31G (d, p) basis sets. It is clear from Fig. 4 that the positive and negative phase is represented in orange and green color respectively. It can be seen from the plots that the region of HOMO and LUMO levels spread over the free anthraquinones (M1-M5). However, the HOMO and the LUMO orbitals are located all over the anthraquinone ring plane of anthraquinone glucosides rather than the glucose ring. This indicated that the atom with more occupation of LUMO should have ability to gain an electron (Mezzina et al., 2021). However, the hydroxyl oxygen atoms in the glucose ring act as electron donors and easily form hydrogen bonding interactions with the surrounding amino acids, resulting in stronger ability to detach electrons. It is interesting for M9 to find that the glucose ring donates electrons, while the anthraquinone ring plane accepts electrons. From the energy levels of HOMO and LUMO orbital for these anthraquinone compounds at the active site (Table 2), it can be seen that the energy gap of HOMO-LUMO is the highest for M9 (0.148ev), which reflects the chemical stability of the molecule.
Due to the molecular resistance can change or deform the number of electrons, chemical hardness (η) is associated with the stability and reactivity of a chemical system (Adejumo et al., 2020). The combined anthraquinone M9 has the highest ∆Egap, which shows a stable molecule and it needs much excitation energy to reach the manifolds of excited states (Huang et al., 2017). As can be seen, the calculations electrophilicity (ω) performed at anthraquinone glucosides are very close to each other when compared with the anthraquinones. The other important electronic property dipole moment values for the molecules are also given in Table 2. Generally, the combined anthraquinone glucosides have larger dipole moments than that of the free anthraquinones.
3.5 Topological properties analysis
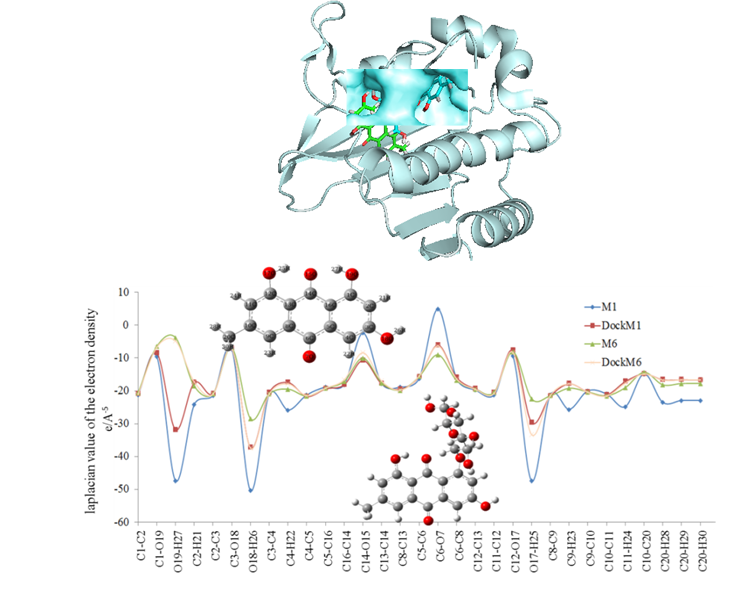
In general, the charge concentration/depletion at the BCP (bond critical point) of the chemical bond provides interesting information about the chemical bonds by using the Laplacian of electron density ∇2ρbcp (Wang et al., 2016; Prabavathi and Nilufer, 2015). Due to emodin (M1) and its anthraquinone glucoside emodin-8-beta- glucosicide (M6) have the lowest binding energies among the structural analogues, we paid attention to the change of their electron densities of the chemical bonds. Based on the Multiwfn program, the (3,-1) type of BCP for all chemical bonds of M1 and M6 were evaluated for both two status. Based on further topological analysis, the electron density (ρbcp) and their Laplacian values (∇2ρbcp) are listed in Supplementary Table S2. The (3,-1) type of BCP for all chemical bonds were found by the Multiwfn program suggested the existence of the covalent interactions (Lu and Chen, 2012). It can be seen from Supplementary Table S3 that the electron densities of the molecule M6 at active site are very close to the optimized phase.
However, the electron densities of the M1 at active site are higher than that of the other status. The electron densities (ρbcp) of the C-C bonds on aromatic ring for these molecules in both forms are ranged from 1.583 to 2.159 eÅ-3, implying strong covalent interactions (Luis, 2016). Due to the strong electrostatic interactions between the carbonyl O7 atom and amino acids Asn46, His99, Phe104 and Val118, the electron density of the bond C6-O7 remains the highest value. In short, almost all of the electron densities of M1 at the active site are reduced, while the M6 remains constant (Fig. 5). On compared with other chemical bonds, the electron densities of all O-H and C-O bonds are generally higher while the corresponding Laplacian values are lower except C6-O7, which suggests that the charge densities at the BCP are highly concentrated.
On the other hand, low value of electron density and positive character of the Laplacian for M1 are the evidences for the covalently rupture of C6-O7 bond. Moreover, a change in the sign of the Laplacian from the positive values (4.952 eÅ-5) to a negative character at the active site (-6.037 eÅ-5), indicates the depletion of the charge density confirming the C6-O7 bond of M1 cleavage while interacting with the DNA gyrase B. Once glucoside ring is introduced into the M1 (Fig. 5), low value of electron density ρ(r) and positive data of the Laplacian of the O19-H27 are the evidences of the highly polarized and covalently decreased bonding interaction (Luis, 2016). Actually, most of the Laplacian values of electron densities are reduced at the active site. Among which, the highest electron density values (2.766-2.788 eÅ-3) can be found on the carbonyl C-O bonds of the parent ring in both situations. The electronic Laplace values are all negative for M1 molecule except the positive values of C14-O15 and C6-O7 on the anthraquinone ring plane, implying a more concentrated character at the active site. The electron densities of the three hydroxyl groups on the M1 hydroxyanthraquinone plane become more depleted after interacting with the DNA gyrase B, implying strong hydrogen bonding interactions with the surrounding amino acids Gly77, Gly101and Asp25 (Renuga et al., 2015). Most of the covalent bonds of the M6 molecule become more aggregated, which may be related to a more stable structure at the active site. However, positive character of the Laplacian values are the evidences for the covalently rupture of C14-O15 and C6-O7. This is described to the strong electrostatic interactions with the surrounding polar amino acids Arg136, Glu137, Gly164, Val118, Val97 and His99 etc.
{kind=link}