Animals
All animal procedures were reviewed and approved by the University Committee on Animal Resources of the University of Rochester Medical Center for compliance with federal regulations prior to the initiation of the study. Tg(APPswe,PSEN1dE9)85Dbo male mice were obtained from Jackson Laboratories (43832-JAX, C57Bl/6J background) and bred to female C57Bl/6J mice to produce a total of 38 APP positive mice and 29 nontransgenic mice. All mice were co-housed in cages of up to five mice and provided food and water ad libitum. For viral vector validation, 16 12-week-old C57Bl/6J male mice were used.
Adeno-associated viral constructs
Gas6(Myc-DDK-tagged)-mouse growth arrest specific 6 (Gas6) cDNA was purchased from OriGene (Rockville, MD) and used to construct an AAV-packaging vector using In-Fusion HD Cloning Kit (Clontech Laboratories). The cloning vector was packaged into AAV-1 under the SYN1 promoter at the National Institute of Drug Abuse at the NIH by Brandon Harvey, as described (Howard & Harvey, 2017). A deletion mutant lacking the Gas6 Gla and EGF domains, as described in Geng et al., 2017, was created and processed as above.
Viral vector validation study
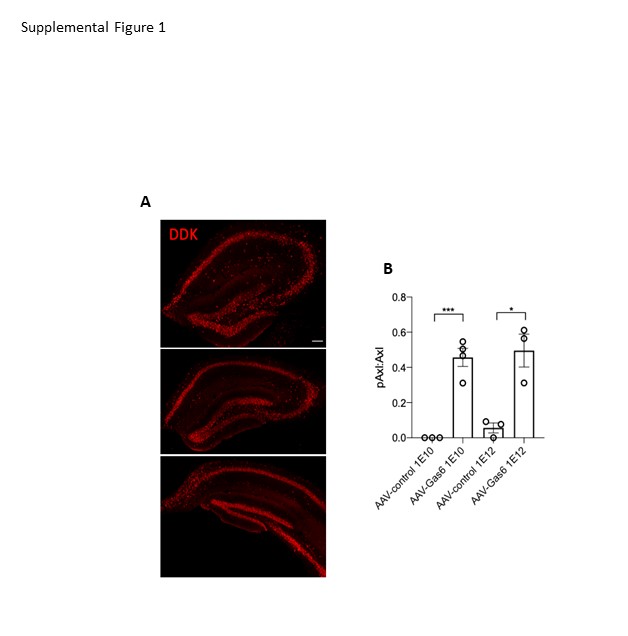
Sixteen 12-week male C57Bl/6J mice received either 1.5 mL of 10E10 vg/mL or 10E12 vg/mL via stereotactic hippocampal injection as described below. Mice were sacrificed four weeks later. Half brains were processed for immunohistochemistry and the other half was processed for western blot, as described below.
Stereotactic injections
The following protocol was used for all viral vector injections: mice were injected with slow-release buprenorphine (0.5 mg/kg, i.p.) and anesthetized with 1.75% isoflurane in 30% oxygen and 70% nitrogen. The head was secured in a Kopf stereotactic apparatus using ear bars and a head holder. Ophthalmic ointment was applied to prevent eye dryness. Hair was removed on top of the skull and the scalp was disinfected with betadine and ethanol prior to incision with a scalpel. Two 0.5 mm burr holes were drilled in the skull at 1.8 mm caudal and +/- 1.8 mm lateral relative to bregma. A 33 GA needle was lowered over the course of two minutes to a depth of 1.5 mm. A Micro-1 microsyringe pump controller (World Precision Instruments, Sarasota, FL) was used to inject 1.5 ml of AAV-Gas6 or AAV-control (1E12 vg/mL, or 1E10 vg/mL for viral vector validation study) over ten min, followed by a five min rest period. After the injection, the needle was raised over the course of two minutes and the protocol was repeated on the opposite side of the brain. After injections were complete, the burr holes were sealed with Ethicon bone wax and the incision was closed with tissue adhesive. Mice recovered in a heated area before being placed in their home cage. Aliquots of AAV-Gas6 or AAV-control were randomized into microfuge tubes by another laboratory member so that the investigator was blinded to what treatment group each mouse was in until the end of data collection. The order of injections were also randomized to control for time of day effects. Mice were monitored for total recovery for three days. All 77 mice were injected over the course of one week at 9 months of age. For the viral vector validation study, 16 12-week-old male mice were injected over the course of two days.
Behavioral assays
Two weeks before all behavioral assays, mice were transferred to a reverse light dark cycle room so that behavioral assays could be run during the awake cycle. Four APP/PS1 male mice were eliminated from novel object and fear conditioning behavioral analyses (one in the control-treated group and three in the Gas6-treated group) due to observation of seizures during behavioral testing. The incidence of observed seizure behavior was 14% in the control-treated male APP group (1/7) and 23% in the Gas6-treated male APP group (3/13); no seizures were observed in the wild type control mice.
Open Field
To assess activity level and anxiety, an open field test was performed. Mice were allowed to explore freely in a 31x31cm box for five min. AnyMaze software (Stoelting, Wood Dale, IL) was used to track mice and the periphery of box was defined by 5 cm from the edge of the box. Distance, mean speed, time spent freezing, and time in periphery was automatically determined using AnyMaze using the center of the mouse’s body. Boxes were cleaned thoroughly with ethanol between animals and male animals were run before females.
Novel object recognition
During the habituation phase, mice were allowed to explore a 31 x 31 cm box for 5 min containing two identical objects spaced ~15 cm apart. All objects used were ceramic doorknobs of 5-6 cm in height and ~3 cm in width. Objects and chambers were washed with 70% ethanol before each trial. Two hours after the habituation phase, each mouse was returned to the experimental cage containing the object to which it was previously exposed (familiar object) as well as a novel object. Placement of the novel object was randomized for each mouse. Mice were allowed to explore familiar and novel objects during a 5 min test that was videotaped for subsequent analysis using the AnyMaze software. Scoring of NOR performance was based on time spent exploring both familiar and novel objects. The behavior of the mouse was considered exploratory when the animal’s head faced the object with the neck extended. Simple proximity, passing-by, or standing on the object did not count as exploration. Mice that spent less than 20 seconds exploring both objects during either the training or testing phase were not included in the analysis (1 APP/PS1 AAV-control male mouse, 3 APP/PS1 AAV-Gas6 male mice, 1 wild type AAV-Gas6 male mouse, 1 APP/PS1 AAV-control female mouse, 1 wild type AAV-control female mouse, and 1 wild type AAV-Gas6 female mouse). Discrimination index was defined as (Time with NO - Time with FO)/(Time with NO + Time with FO).
Contextual fear conditioning (CFC)
Mice underwent cued and contextual fear conditioning, as previously described (Matousek et al., 2012). Briefly, on conditioning day, mice were allowed to explore the conditioned context, which consisted of a Plexiglas chamber and metal floor grid (model H10-11M; Coulbourn Instruments, Whitehall, PA, USA). After 3 min, 15 sec of white noise (80 dB) was presented co-terminating with a 2 s, 0.75 mA foot shock. This noise-shock pairing was repeated twice for a total of 3 shocks with a 30 sec interval between shocks. Twenty-four hours later, mice were re-exposed to the conditioning chamber for 5 min and freezing responses were measured using FreezeView Software (Coulbourn Instruments) to test contextual long-term memory. Four hours later, mice were placed in a novel context consisting of a 15 cm open-topped plastic cylinder with bedding on the floor for 3 min followed by re-exposure to the white noise for 3 min, to test hippocampal-independent memory. All data were video recorded using FreezeFrame Video-Based Conditioned Fear System and analyzed by FreezeView Software (Coulbourn Instruments). CFC was performed in all male mice before female mice.
Methoxy-04 injection and flow cytometry
After behavioral testing, and 24 h before sacrifice, animals were intraperitoneally injected with Methoxy-04 (Tocris Bioscience) at 4.35 mg/kg. On the day of sacrifice, mice were deeply anesthetized with xylazine (10 mg/kg) and ketamine (100 mg/kg) and perfused intracardially with 0.15M phosphate buffered saline containing 0.5% sodium nitrite (weight/volume) and 2 IU heparin/mL. After perfusion, brains were collected and bisected along the longitudinal fissure. For all animals, one half brain was submerged in 4% paraformaldehyde, pH 7.2 in PBS at 4°C for 24 hours, and processed for immunohistochemistry as described below. For half of the animals, hippocampi from the other half of the brain were dissected and processed for ELISA as described below. For the other half of the animals, each hippocampus from the other half brain was processed for flow cytometry as follows: hippocampi were dissected and dounce homogenized in 3 mL FACS buffer (PBS + 0.5% BSA). Brain homogenates were filtered through a 70 µm filter, rinsed with FACS buffer, and centrifuged at 400xg for 5 min at 4°C. Pellets were resuspended in 40% percoll (GE Healthcare) and centrifuged at 500xg for 30 min without brake. Supernatants were removed and pellets were resuspended in 100 mL FACS buffer containing 1:100 Fc block (2.4T2, BD) and transferred to a 96 well plate. The following antibodies were added: CD45 (30-F11, APC/Cy7 1:400, Biolegend), CD11b (M1/70 Biolegend) and incubated for 30 min at 4°C. Cells were washed once and stained with the 7AAD viability dye (ThermoFisher). They were then sorted on a BD FACSAria II (BD Biosciences) and gated on CD45lo/CD11b+ following exclusion of debris, doublets and dead cells. Cells were sorted into Buffer RLT Plus (Qiagen) + 10 mL/mL beta-mercaptoethanol and stored at -80°C until processing for RNAseq.
Tissue processing and immunohistochemistry
For immunohistochemistry, fixed half brains were equilibrated in 30% sucrose in PBS overnight, frozen in cold isopentane, and stored at -80°C until sectioning into 30 mm sections on a -25°C freezing stage microtome. Free-floating sections were stored in a cryoprotectant solution until assayed. For staining, sections were washed in PBS and blocked with normal donkey or goat serum for one hour at room temperature. Sections were then incubated in primary antibodies (goat anti-Gas6 1:500, R&D Systems AF986-SP; rabbit anti-Iba-1 1:2000, Wako NC9288364; or rat anti-CD68, 1:500, Abcam ab53444), for 48 h at 4°C, after which they were washed and incubated in secondary antibodies (Invitrogen Alexa fluors 488 or 594, 1:2000) for 2 h at room temperature.
Image acquisition and analysis
Slides were imaged on an Axioplan Iii (Carl Zeiss, Oberkochen, Germany) microscope. Slidebook software (Intelligent Imaging Innovations, Denver, CO) was utilized for image acquisition, in which 10x or 40x images were taken of 3-4 sections or 5-6 plaques per mouse. Slides were labeled with blinded animal numbers. Images were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD). For plaque % area and particle analysis, ROIs were drawn using the bottom leaflet of the dentate gyrus and upper CA1 field as anatomical guidelines. Images where then thresholded using ImageJ’s “Otsu” algorithm and % area and particle statistics were measured. For CD68 and Iba-1 quantification, ROIs were drawn around each plaque and enlarged such that each ROI encompassed the 15 mm surrounding each plaque. Images were thresholded using the “Otsu” algorithm. % area and integrated density were measured.
Western blot
For the viral vector validation study, dissected hippocampi were flash frozen in ice cold isopentane and stored at -80°C. Frozen hippocampi were weighed and homogenized in T-PER (Thermo Scientific, 50 mg/mL) for 30 s, vortexed, and sonicated for 30 s. Protein concentration was determined using BCA assay (Thermo Scientific). Protein was then diluted in 2x sample buffer (125 mM Tris-HCl; 4% SDS; 20% glycerol) at a concentration of 1 mg/mL. Samples were briefly vortexed, boiled for 10 min, and 15 mL was electrophoresed on a Tris-HCL polyacrylamide gel. Gels were transferred to a 0.4 mm PVDF membrane at room temperature overnight. Membranes were washed and blocked in 5% BSA/TBS-T for one hour at room temperature. After washing in TBS-T, membranes were incubated in pAxl (Invitrogen PA5-64862) at 1:100 overnight at 4°C. Membranes were then washed and incubated in peroxidase-linked secondary antibodies (Invitrogen) for one hour at room temperature. Blots were treated with ECL substrate (Supersignal West Dura Kit, Thermo Scientific) and bands were visualized using an Azure c600 Gel Imaging System (Azure Biosystems, Dublin, CA). Blots were stripped for 10 min in Strip Reblot Plus Strong Solution (Miltenyi). Blots were then re-probed with mouse anti-Axl (R&D) at 1:100 and protocol above was repeated.
ELISA
Hippocampal lysates from Gas6 and control-treated APP/PS1 mice were centrifuged at 100,000 g for 1 h at 4°C to separate soluble aggregations of Aβ (monomers and oligomers) from large, insoluble Aβ fibrils. Supernatants were collected as the soluble fragments. CXCL13 and CCL2 chemokine levels were measured utilizing respective mouse ELISA kits (R&D MCX130 and MJE00B). Soluble samples were diluted 1:1 in kit buffer. Plates were read with Microplate Absorbance Reader (Bio-Rad). Linear regression models were used for CXCL13 & CCL2.
RNAseq
Total RNA was isolated using the RNeasy Plus Micro Kit (Qiagen, Valencia, CA). RNA concentration was determined with the NanoDrop 1000 spectrophotometer (NanoDrop, Wilmington, DE) and RNA quality assessed with the Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA). One ng of total RNA was pre-amplified with the SMARTer Ultra Low Input kit v4 (Clontech, Mountain View, CA) per manufacturer’s recommendations. The quantity and quality of the subsequent cDNA was determined using the Qubit Fluorometer (Life Technologies, Carlsbad, CA) and the Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA). 150 pg of cDNA was used to generate Illumina compatible sequencing libraries with the NexteraXT library preparation kit (Illumina, San Diego, CA) per manufacturer’s protocols. The amplified libraries were hybridized to the Illumina flow cell and sequenced using the NovaSeq6000 sequencer (Illumina, San Diego, CA). Single end reads of 100 nt were generated for each sample. Two to six biological replicates were sequenced for each group.
Bioinformatic analyses
The RNAseq bam file was mapped to mm10 reference genome obtained from Ensemble with STAR (Dobin et al., 2013). featureCounts was used to assign reads to genomic features (Liao et al., 2014). DESeq2 was used to perform differential expression analysis (Love et al., 2014). Groups compared were AAV-Gas6-treated APP and AAV-control-treated APP. Genes were considered differentially expressed if padj<0.05. Differentially expressed genes were separated based on the direction of their fold change and gene ontology analyses on the resulting lists of genese were conducted with clusterProfiler package (Yu et al., 2012).
Primary microglial isolation
C57BL/6J Axl+/− and Axl−/− pups (Jackson labs: Axltm1Grl/J, generously donated from the Calvi Lab at the University of Rochester) ages P0-P2 were sprayed with 70% ethanol and decapitated. Brains and meninges were removed. Tissue was washed with ice cold HBSS three times. 1 ml of 0.25% trypsin + 50 µL DNase was added and incubated at room temperature for 7 min. Tissue was again washed twice with HBSS. Tissue was dissociated using a P1000 pipettor by pipetting up and down until tissue was fully dissociated, then was filtered through a 70 µm nylon cell strainer into a new tube. DMEM + 10% FBS + 1% pen/strep was added and samples were spun at 300 x g for 10 minutes. Samples were then transferred to vented T-75 flasks. Media was changed after 24 h after which cells were left to incubate for two weeks. At this time, microglia were isolated from astrocytes using a mild trypsinization protocol: cells were rinsed with DMEM without FBS, and incubated in 1x trypsin/EDTA (Invitrogen 25200-056) diluted 1:3 with DMEM without FBS and incubated at 37°C until the astrocytic layer was fully removed from the microglial layer (20-40 min) (Saura et al., 2003). Microglia were then removed with undiluted 0.25% trypsin, collected, spun at 300g for 10 min, and plated into cell-culture treated 96 well plates with serum-free media for 24 hours before treatment. Cells were then treated with beta-amyloid (1–42) HiLyte Fluor 488 (AnaSpec) with or without 50 nM rmGas6 (R&D Systems) for 15 min, 30 min, 60 min, or 120 min. Cells were then washed and collected for flow cytometry, where they were analyzed using an LSR II (Becton Dickinson) in the University of Rochester Medical Center Flow Cytometry Core. Data was acquired using FCS Express 7 (De Novo Software, Pasadena, CA).
Statistics and data analysis
All statistical comparisons, except for those pertinent to RNAseq data, were performed with Prism (GraphPad Software, version 8, San Diego, CA) using unpaired Student’s t-tests, one-way ANOVAs, and two-way ANOVAs, as required based number of experimental vs control conditions. The Shapiro-Wilk test was used to determine normality of the data. Based on results of normality test, Student’s t-test or Mann Whitney test were employed when two group means were compared. Tukey’s correction for multiple comparisons was conducted where appropriate. All results are expressed as mean +/- SEM. A p-value of <0.05 was considered significant in all experiments.
{kind=link}