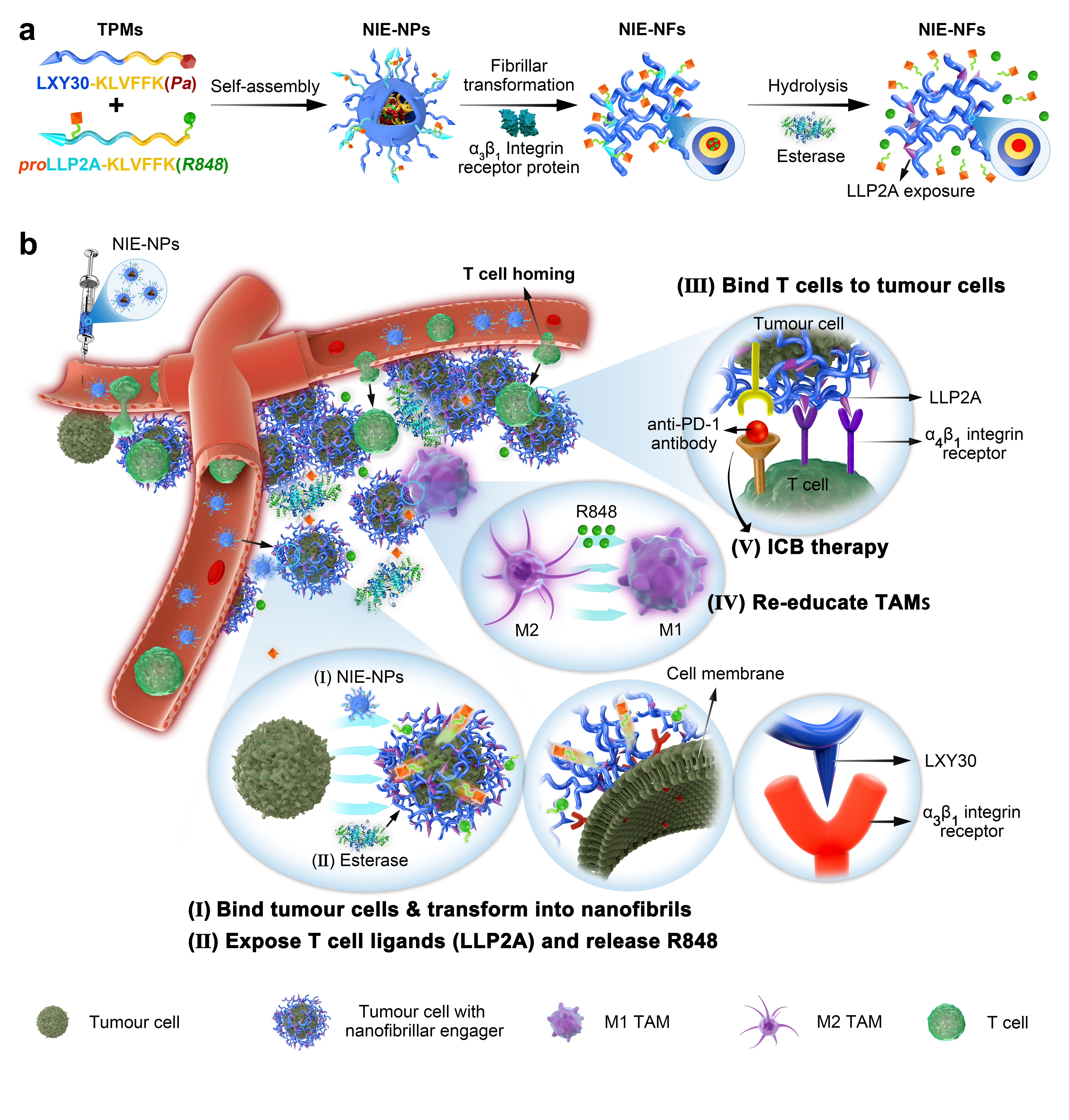
Self-assembly, fibrillar transformation and esterase hydrolysis of programmable bispecific NIE
Two transformable peptide monomers (TPM1 and TPM2) were synthesized and characterized (Fig. 1a and Supplementary Fig. 1). As the proportion of water in the mixed solvent (water and DMSO) of the TPM1 and TPM2 mixture solution (the ratio of 1:1) was increased, there was a gradual decrease in fluorescence peak at 675 nm due to the aggregation-caused quenching (ACQ) properties of Pa dye (Fig. 1b), reflecting the gradual formation of NIE-NPs via self-assembly. Concomitantly, there was a modest decrease in the absorption peak at both 405 and 680 nm (Supplementary Fig. 2a). Nanoparticles were analyzed by transmission electron microscopy (TEM) and dynamic light scattering (DLS). TPM1 and TPM2 each alone were able to self-assemble to form nanoparticles (NPsTPM1 and NPsTPM2) at 18 and 55 nm, respectively. NIE-NPs, assembled from 1:1 mix of TPM1 and TPM2 into nanoparticles at around 28 nm, which fell between the sizes of NPsTPM1 and NPsTPM2 (Supplementary Fig. 2b). The critical aggregation concentration (CAC) of NIE-NPs was determined to be 8 µM (Supplementary Fig. 2c). We have also demonstrated that NIE-NPs could maintain good serum stability and proteolytic stability over 7 days at 37 oC (Supplementary Fig. 2d).
To verify the receptor-mediated fibrillar transformable process of NIE-NPs in vitro, soluble α3β1 integrin protein (receptor for LXY30) was added to NIE-NPs solution. After 24 h of incubation at room temperature, a fibrillar network (NIE-NFs, width diameter about 8 nm) with a broad size distribution was clearly detected (Fig. 1c,f). No transformation was observed in the NIE-NPs preparation without the addition of α3β1 integrin protein, even after 24 h (Supplementary Fig. 2e). The CAC of NIE-NFs was determined to be 5 µM, which was lower than that of NIE-NPs (8 µM), indicating that NIE-NFs has higher propensity to form nanostructures than NIE-NPs (Supplementary Fig. 2f). The process of NIE-NPs transformation could be monitored by the fluorescent intensity of Pa (Fig. 1d). Addition of α3β1 integrin protein triggered a gradual decrease in fluorescence intensity of Pa over time, reflecting an increase in fluorescence quenching in NFs1, indicating that the packing of Pa in NIE-NFs was denser. In addition, a 5 nm fluorescence red shift (from 675 nm to 680 nm) with fluorescence peak reversal (from 680 nm to 725 nm) were observed, indicating that the disordered arrangement of Pa in the core of NIE-NPs was transformed into J-aggregate form in NIE-NFs.30-32 We have also investigated the responsiveness of proLLP2A displayed on the NIE-NPs surface to soluble α4β1 integrin protein in the presence and absence of esterase (Fig. 1e,f and Supplementary Fig. 3). Soluble α4β1 integrin protein alone was not able to alter the structure of NIE-NPs displaying proLLP2A, even after 24 h of incubation. In contrast, successive addition of esterase, followed by soluble α4β1 integrin protein was able to elicit conversion of NIE-NPs to fibrillar network after 24 h of incubation. This expected result confirmed that esterase was able to convert pro-ligand proLLP2A to ligand LLP2A, which in turn was able to trigger receptor-mediated transformation of NIE-NPs to NIE-NFs. We also monitored the conversion of proLLP2A ligand to LLP2A ligand by HPLC. As shown in Supplementary Fig. 4, majority of proLLP2A were found to be converted to LLP2A after incubation with porcine liver esterase for 8 h at pH 7.4 and 37 oC.
Circular dichroism (CD) spectroscopic analysis of the transformation process of NIE-NPs showed a gradual progression of a negative signal at 216 nm and a positive signal at 195 nm upon incubation with α3β1 integrin protein or combination esterase/α4β1 integrin protein, indicative of β-sheet formation (Fig. 1g) and consistent with TEM results shown in Fig. 1c,e.23, 33 In vitro release behaviour of R848 from NIE-NFs was studied at pH 6.5 with addition of esterase to simulate TME condition. As shown in Fig. 1h, about 45% of R848 was released in the first 24 h, after which the release rate gradually slowed down and about 86% cumulative release was observed by 168 h, indicating that prolonged and sustained release of R848 could occur at the TME, which may be due to the slow hydrolysis of the ester bond to R848 within the fibrillar structure. To demonstrate the unique transformable property of NIE-NPs, we designed a related negative control nano-immuno-engager nanoparticle (CNIE-NP) formed by assembly of two negative control TPMs without b-sheet forming KLVFF peptide sequence, at a ratio of 1:1 (CTPM3: LXY30-KAAGGK(Pa) and CTPM4: proLLP2A-KAAGGK(R848), Supplementary Fig. 1). As expected, α3β1 integrin protein was unable to transform CNIE-NPs to fibrillar structures even after 24 h, indicating that the b-sheet peptide was required for the transformation of NIE-NPs to NIE-NFs (Supplementary Fig. 5a,b). CD spectroscopic analysis of CNIE-NPs remained the same with or without the addition of α3β1 integrin protein, indicating that β-sheet was not formed (Supplementary Fig. 5c).
In vitro evaluation of bispecific NIE facilitating Teff cells homing and reeducating macrophages
We investigated the binding affinity and selectivity of LXY30 to a3b1 integrin and LLP2A to a4b1 integrin via flow cytometry. To ensure the specificity and accuracy of binding analysis, K562 human myeloid leukemia cell line stably transfected with a3b1 or a4b1 integrins were used. As shown in Supplementary Fig. 6a, biotinylated LXY30 ligand displayed little affinity for K562 (a4b1+) cells, whereas biotinylated LLP2A ligand showed high affinity for these cells. As expected, proLLP2A ligand showed no significant affinity to these a4b1 integrin expressing cells, but the affinity was restored after proLLP2A ligand was treated with esterase (Supplementary Fig. 6b). We also analyzed the binding affinity of LXY30 and LLP2A ligand to K562 (a3b1+) cells. As shown in Supplementary Fig. 6c, LXY30, but not LLP2A, was found to have high affinity against these a3b1 integrins expressing cells. To further characterize the interaction between NIE-NPs and α3β1 integrin receptors on the surface of living cells, we chose α3β1 integrin expressing 4T1 murine breast cancer cell. As expected, flow cytometry analysis confirmed that LXY30, the high affinity α3β1 integrin ligand, did bind to 4T1 tumour cells (Supplementary Fig. 7). We have also found that NIE-NPs was slightly cytotoxic against 4T1 tumour cells, with 84.5% cell viability at 50 mM (Supplementary Fig. 8). We investigated the distribution of nanoparticles by tracking the red fluorescent signal emitted by Pa using confocal laser scanning microscopy (CLSM). Six hours after incubation of 4T1 tumour cells with NIE-NPs, a strong red fluorescence signal was observed on the cell surface and its vicinity but not inside the cells (Fig. 2a). In contrast, the fluorescent signal of Pa in CNIE-NPs-treated group was found to be concentrated primarily in the cytoplasm of the cells. To study the retention and stability of formed nanofibrillar network on the surface of tumour cells, unbound nanoparticles were washed off after 6 h of incubation and fresh medium without nanoparticles was added to incubate cells for another 18 h. As expected, NIE-NPs treated cells still retained strong red fluorescence signals on the cell surface at 24 h, displaying that the capacity of prolonged retention of bispecific NIE (Fig. 2b). In sharp contrast, only weak fluorescence signal was observed inside the cells treated with CNIE-NPs after 24 h. This is probably due to the enzymatic degradation of the already endocytosed CNIE-NPs after 18 h of incubation, but without any new endocytic uptake during that time period. TEM images confirmed the presence of nanofibrillar network on the surface of, and between 4T1 tumour cells after incubation with NIE-NPs for 24 h, but absence of such nanofibrillar structures on cells treated with CNIE-NPs (Fig. 2c). The fibrillar structures further away from the tumour cell surface were probably induced by the secreted tumour exosomes displaying α3β1 integrin proteins.16, 34 We also investigated the effect of esterase on the interactions between NIE-NPs and T-cell surface a4b1 integrin, after converting pro-ligand pro-LLP2A to LLP2A displayed on the surface of NIE-NPs. Live GFP transfected Jurkat T-lymphoid leukemia cells with high expression level of constitutively activated a4b1 integrin protein were used to mimic T cells. As shown in Fig. 2d, after 6 h incubation of Jurkat cells with NIE-NPs (pre-treated with esterase), a luxuriant red fluorescent layer was found surrounding the Jurkat cells, indicating that the conversion of pro-ligand to LLP2A ligand was successful. Scanning electron microscopy (SEM) confirmed the presence of fibrillar network on the surface of NIE-NPs-treated 4T1 tumour cells and esterase pre-treated NIE-NPs-treated Jurkat cells (Fig. 2e).
To simulate the processes of initial fibrillar transformation of NIE-NPs on the 4T1 tumour cells surface followed by T cell homing, we incubated 4T1 tumour cells with NIE-NPs for 6 h, unbound NIE-NPs were then washed off, followed by addition of fresh medium containing esterase but without NIE-NPs. After 1 h of incubation, Jurkat cells were added and incubated with 4T1 tumour cells for 2 or 4 h. After that, unbound Jurkat cells were gently removed prior to CLSM imaging (Fig. 2f). As expected, a fibrillar structure layer with red fluorescence was detected surrounding 4T1 tumour cells surface, and Jurkat cells (GFP+) were found to interact with the red fluorescent fibrillar network (bispecific NIE) and in close proximity to 4T1 tumour cells, after 2 h of incubation. As the incubation time was increased to 4 h, many more Jurkat cells were found around the 4T1 tumour cells, which was consistent with our notion that nanofibrillar-based bispecific NIE would facilitate the homing of immune cells. SEM imaging provided critical evidence that the bispecific NIE had played a significant role in direct physical contact between 4T1 tumour cells and Jurkat cells through nanofibrillar network (Fig. 2g). In addition to using Jurkat cells as the surrogate T-cells, we also examined the effects of NIE on the killing of 4T1 tumour cells by activated murine CD8 T cells (a4b1 integrins pre-activated by Mn2+). As shown in Supplementary Fig. 9, NIE-NP transformed into nanofibrillar network on the tumour cell surface, and as a result of esterase treatment, the displayed LLP2A captured activated T-cells, leading to about 50% tumour cell kill (Supplementary Fig. 10), confirming that the nanofibrillar network did not dampen immune synapse formation and killing efficiency of CD8 T cells. Similar studies were also performed on the negative control CNIE-NPs, which were taken up by 4T1 tumour cells, instead of forming nanofibrillar network on the cell surface, and as expected, no bound Jurkat cells were detected (Supplementary Fig. 11).
The conversion of TAMs from an immunosuppressive M2-polarized phenotype to an anti-tumourigenic M1-polarized phenotype is one of the major immunotherapeutic strategies for reprograming the immunosuppressive TME. Macrophage polarization states demonstrate hallmark morphology, e.g., elongated projections for M2-like cells as opposed to a round and flattened morphology for M1-like cells (Supplementary Fig. 12). IL-4 has been used to induce bone marrow derived macrophages (BMDM) to M2-polarized macrophages, as reflected by the increase in expression level of the metabolic checkpoint enzyme arginase-1 (Arg1) and mannose receptor-1 (Mrc1). R848 has been reported to be a powerful driver of the M1-phenotypes in vitro, resulting in elevated level of interleukin 12 (IL-12) and nitric oxide synthase (Nos2) produced by these cells.8 Here we investigated the possibility of using NIE-NFs to reeducate macrophages from M2 phenotype to M1 phenotype. Not unexpected, incubation of M2-state macrophages with NIE-NFs, preformed from NIE-NPs with soluble a3b1 integrin protein, did not have significant change in morphology and expression level of Arg1 and Mrc1 even after 12 h (Fig. 2h), which can be explained by the lack of R848 released from NIE-NFs. In contrast, addition of esterase to the culture medium followed by 12 h incubation resulted in morphological change of M2-state macrophages towards M1-state, a decrease in Arg1 and Mrc1, and an increase in IL-12 and Nos2 expression as measured by qPCR. These changes were even more pronounced after 24 h, at which time the macrophages were completely transformed to a round and flattened morphology, with further decrease in Arg1 and Mrc1, and increase in IL-12 and Nos2 expression. We believe the ability of bispecific NIE to anchor on the surface of tumour cells, affording the sustained release of R848 from the fibrillar network, would generate a durable anti-cancer immunoactive TME. Interestingly, we have also found a significant change in macrophage morphology from M2-state towards M1-state after 24 h treatment with CNIE-NPs, which was perhaps because the particulate CNIE-NPs were phagocytosed by M2-macrophages, releasing R848 intracellularly, resulting in conversion of these macrophages from M2 to M1 phenotype (Supplementary Fig. 13).
In vivo evaluation of bispecific NIE targeting tumour cells and nanofibrillar-transformation to facilitate Teff cell homing and activate immunity
NIE-NPs was found to be non-toxic: blood counts, platelets, creatinine and liver function tests obtained from normal Balb/c mice treated with eight consecutive q.o.d. intravenous (i.v.) doses of NIE-NPs (13 mg/kg) were within normal limits (Supplementary Figs. 14 and 15). In vivo blood pharmacokinetics (PK) studies in rats showed that NIE-NPs possessed a long circulation time (T-half (α): 2.866 h and T-half (β): 23.186 h), indicating its stability during circulation (Supplementary Fig. 16). For biodistribution studies, NIE-NPs were tail vein injected once (13 mg/kg) into Balb/c mice bearing syngeneic orthotopic 4T1 breast cancer; 10, 24, 48, 72, 120 and 168 h later, tumour and main organs were excised for ex vivo fluorescent imaging (Fig. 3a,b). Significant fluorescent signal of Pa was found to persist in tumour tissue for over 168 h, while fluorescent signal in normal organs began to decline after 10 h and was almost undetectable in the main organs at 72 h. In sharp contrast, fluorescent signal of Pa at tumour tissue treated by CNIE-NPs was found to gradually decline over time after peaking at 24 h (Fig. 3c). By 168 h, less than 2.91% of the peak fluorescent signal for CNIE-NPs remained in the tumour, whereas for NIE-NPs, over 60.49% signal remained in the tumour (Fig. 3d). We also found that the red fluorescence signals from pheophorbide a (Pa) in NIE-NPs were detected throughout the entire tumour tissue, not just in the tumour periphery (Supplementary Fig. 17). Most of the red fluorescence (Pa) was observed in the extracellular space in close proximity to the tumour cell membranes, as indicated by the green arrows. Prolonged retention of fluorescent signal in NIE-NPs-treated mice could be attributed to in situ receptor-mediated transformation of NIE-NPs into NIE-NFs at the TME. TEM studies on excised tumour sections, 72 h after i.v. administration, showed abundant bundles of nanofibrils in the extracellular matrix while no such nanofibrils were observed in negative CNIE-NPs-treated and untreated mice (Fig. 3e). Fluorescent micrographs of tumour and overlying skin revealed intense fluorescent signal in tumour region but negligible signal in normal skin (Fig. 3f). This is consistent with our notion that (1) NIE-NPs would leak into the TME through leaky tumour vasculatures (EPR effect), followed by interaction with a3b1 integrin on tumour cells and tumour associated exosomes to generate NIE-NFs, and (2) blood vessels are not leaky in normal skin. The tumour tissue distribution of R848 over time was also determined with high pressure liquid chromatography-mass spectroscopy (HPLC-MS, Fig. 3g). We found R848 uptake by tumour was quite high at 24 h (3.62 μg per g tissue), and about one-third of R848 was found to retain at the tumour site (1.14 μg per g tissue) even at 7 days after injection of NIE-NPs. Although CNIE-NPs could also deliver significant amount of R848 to the tumour site (83% of what NIE-NPs could deliver), retention of R848 at the tumour site was much lower than that of NIE-NPs. Prolonged retention and release of R848 in tumour site indicates that a sustained immune-active TME could be achieved with NIE.
To evaluate if the bispecific NIE displaying LLP2A and R848 at the TME could facilitate Teff cells homing to the tumour sites and reeducate TAMs in vivo, tumours from NIE-NPs-treated mice were excised on day 15 after a single i.v. injection of NIE-NPs (13 mg/kg), and the immune cell populations within the tumours were analyzed by flow cytometry, immunohistochemistry (IHC) and qPCR. Experiment using CNIE-NPs as an untransformable/endocytic negative control group was also performed at the same time. We found that tail-vein injection of NIE-NPs resulted in greatly increase concentration ratio of cytotoxic T cells and a sustained immunoactive TME. First, NIE-NPs was found to significantly stimulate the production of chemokine CXCL10 at the tumour site (Fig. 3h), which was known to facilitate Teff cells recruitment.35, 36 This should be attributed to R848 which is known to induce the production of chemokines.37, 38 R848 covalently linked to NIE-NPs via an ester bond was released from the nanofibrillar network anchored at the TME by endogenous esterases. As a result, released R848 would interact with a variety of cells in the TME, such as tumour cells, endothelial cells, and some immune cell subsets (dendritic cells, monocytes, macrophages, and B cells) to produce a range of chemokines and cytokines. Indeed, we observed that the proportion of CD45+CD3+ and CD45+CD3+CD8+ T cells in the NIE-NPs-treated tumour tissue was substantially higher than those from mice treated with endocytic CNIE-NPs or saline (Fig. 3i, Supplementary Fig. 18 and 19a). More specifically, the percentage of CD3+CD8+ T effector cells in tumours was found to be 18 and 4-fold increase, relative to that of saline and CNIE-NPs-treated mice, respectively.
Second, we found that the relative abundance of CD4+Foxp3+ Tregs at the tumour site was substantially lower in mice that received NIE-NPs treatment than those in mice treated with CNIE-NPs, i.e. (4.97% versus 13.0%) or saline (4.97% versus 14.6%). The ratio of tumour-infiltrating CD8+ killer T cells to immunosuppressive Tregs (CD3+CD4+Foxp3+), which could be an indicator of anti-tumour immune balance, was found to be the highest in NIE-NPs treated group. We also analyzed the expression (CD49+) and activation (CD29+) of a4b1 integrin in tumour-infiltrating and blood circulating CD8 and CD4 T cells (Fig. 3i and Supplementary Fig. 19b,c). Although both blood circulating and tumour infiltrating CD8 and CD4 T cells in mice bearing 4T1 tumour were found to be both CD49+ and CD29+, their expression levels were higher in the tumour infiltrating cell populations. This may be because there was more chemokine and cytokine secretion in tumour tissues, therefore inducing more a4b1 integrin expression and activation. More excitingly, we found that after NIE-NPs treatment, there were more CD8+CD49+ T cells and CD8+CD29+ T cells in both circulating and tumour infiltrating cell populations. This data indicates that NIE-NP treatment, probably via chemokine induction, could promote CD49 expression and CD29 activation of T cells, and therefore homing and retention of these cells by the displayed LLP2A at the TME. We also evaluated T cell activation and exhaustion makers (CD69 and LAG-3) in tumour tissues, which could better access the killing effect of T cells against 4T1 tumour cells (Supplementary Fig. 20). Compared to CNIE-NPs and saline treated groups, mice treated with NIE-NPs exhibited higher CD69 expression and lower LAG-3 (an immune check point receptor) expression, the desirable anti-tumour immune phenotype of the TME. IHC staining of tumour tissue sections also confirmed an increase in CD8/CD4 and decrease in Foxp3 positive Treg cells (Fig. 3j).
Third, IHC staining of tumour sections demonstrated an increase in M1-polarized macrophage marker CD68 and a decrease in M2-polarized macrophage marker CD163 in the NIE-NPs treated group, compared to the tumour tissue excised from mice treated by CNIE-NPs. This could be explained by the sustained release of R848 at the tumour site, causing the phenotypic reeducation of TAMs. Fourth, gene expression level of cellular immune related markers (IFN-g, TGF-b) and macrophage markers (IL-12, IL-10, Nos2 and Arg-1) were also evaluated by qPCR. As shown in Fig. 3k, the high expression level of IFN-γ and low expression level of TGF-b in the tumour tissue confirmed that a strong tumour-specific immune response had been elicited. Furthermore, the secretion of IL-12 and Nos2 was found to be significantly upregulated, while the secretion of IL-10 and Arg-1 was significantly down-regulated, indicating a significant phenotypic conversion of TAMs from M2 state to M1 state, with NIE-NPs treatment, but not CNIE-NPs treatment nor saline control.
Intrinsic anti-tumour immune efficacy of bispecific NIE
Therapeutic efficacy study of programmable bispecific NIE was performed in syngeneic orthotopic 4T1 breast cancer-bearing mice. Mice were randomly divided into six groups, each received a different treatment regimen: (1) Saline; (2) (EK)3-KLVFFK(Pa)/(EK)3-KLVFFK(R848); (3) proLLP2A-KLVFFK(R848) (single monomer); (4) LXY30-KAAGGK(Pa)/proLLP2A-KAAGGK(R848) (untransformable negative control CNIE-NPs); (5) LXY30-KLVFFK(Pa)/proLLP2A-KLVFFK(Pa) (fibrillar-transformation but absence of R848); (6) LXY30-KLVFFK(Pa)/proLLP2A-KLVFFK(R848). Regimen 6 is the complete NIE-NPs, containing all 4 critical components: LXY30, proLLP2A, R848, and KLVFF, whereas regimen 2, 3, 4 or 5 all lack some components of NIE-NPs. When tumour volume reached about 50 mm3, all treatment regimens were tail vein injected consecutively eight times q.o.d. (13 mg/kg each dose) and the mice were continuously observed for 21 days (Fig. 4a). As shown in Fig. 4b, regimens 2, 3 and 4 were inactive. Regimen 5 (fibrillar-transformation but no R848) demonstrated significant tumour suppression compared to group 2, 3 and 4. Regimen 6 (NIE-NPs) was found to be the most efficacious with significant tumour growth suppression (Fig. 4b) and prolonged survival (Fig. 4d), indicating the importance of combination T cells homing strategy and sustained release of TLR7/8 agonist. None of the mice in this therapeutic study showed any symptoms of dehydration nor significant body weight loss during the entire treatment period (Fig. 4c). The survival curves correlated well with tumour growth results. The mice treated by regimen 6 (NIE-NPs) achieved a longer median survival time (62 d) compared with other treatment groups (29, 32.5, 33.5, 33.5 and 39 d for regimen 1, 2, 3, 4, and 5, respectively).
To elucidate the mechanism of immunotherapeutic effects induced by bispecific NIE, we collected the tumour tissues and thoroughly evaluate the profile of tumour-infiltration immune cells via flow cytometry analysis (Supplementary Fig. 21). We found that among the six treatment regimens, NIE-NPs (regimen 6) was the most potent inducer for increase in cell numbers of the following cell populations: CD45+, T cells, macrophages, dendritic cells, neutrophiles, B cells and monocytes. Compared to saline control, NK cells and eosinophil cells numbers were also increased with NIE-NPs treatment, but regimens 4 and 5 were equally effective in increasing these cell populations. Additional analysis (Fig. 4e and Supplementary Fig. 22) showed that only the treatment regimens capable of in situ fibrillar transformation and LLP2A display (regimen 5 and 6) were able to greatly increase the frequency of CD3+ and CD8+ T cells within the tumours, particularly in combination with immune adjuvant R848 (regimen 6, NIE-NPs), which was consistent with the observed strongest anti-tumour effects in NIE-NPs. The percentage of M1-phenotype macrophage in total macrophages was 60%, which was much higher than that of other control groups (Supplementary Fig. 23). Tumour sections (H&E) obtained from mice treated with NIE-NPs revealed a marked decrease in Ki-67 expression, an increase in CD8+ T cells, and a decrease in Foxp3 (Treg cells), compared with other control groups (Supplementary Fig. 24). There was an increase in CD68 and a decrease in CD163, indicating that the phenotype of macrophages was reversed after 8 doses of NIE-NPs. It is known that CD8+ T cells secrete cytokines IFN-g and TNF-a to kill tumour cells.15, 39 The expression levels of IFN-g and TNF-a in the tumour tissue were further evaluated by qPCR. As shown in Fig. 4f, treatment regimen 6 (NIE-NPs) was the most efficacious in restoring the immunoactive state of the tumour microenvironment, with the highest expression levels of IFN-g and TNF-a. In addition, NIE-NPs also significantly induced expression of IL-12, IL-6 and Nos2, and suppressed expression of TGF-b, IL-10 and Arg-1, leading to the suppression of the Treg cells recruitment and reeducation of M2-like macrophages to M1 phenotype. To better clarify the contribution of CD8 T cell recruitment to the tumour in NIE-NPs immunotherapy, we performed an CD8 T cell depletion experiment with an antibody (Fig. 4g). Syngeneic orthotopic 4T1 breast cancer-bearing mice were randomly divided into three groups: (1) saline; (2) regimen 6 (NIE-NPs) plus anti-CD8; (3) regimen 6 (NIE-NPs). Anti-CD8 depletion antibody (i.p. injection) was given at an initial dose of 200 μg 3 days before treatment and then given four doses on day 1, 5, 9 and 13. When tumour volume reached about 100 mm3, NIE-NPs were injected via tail vein consecutively eight times q.o.d. (13 mg/kg each dose) and the mice were continuously observed for 21 days. As expected, the therapeutic efficacy of NIE-NPs was greatly diminished in mice given CD8 T cell-depleting antibody (lesser tumour inhibition and shorter survival), confirming the importance of tumour homing of CD8 T cells in the effectiveness of NIE-NP immunotherapy.
Bispecific NIE enhances ICB therapy against breast cancer and lung cancer
Since the receptor-mediated bispecific NIE could significantly mount systemic anti-tumour response by facilitating Teff cells homing and reprogramming of TME, we believe it could enhance the efficacy of ICB therapy. It is well known that tumour cells hijack PD-1 receptors of T cells by overexpression of PD-L1, which can activate PD-1, leading to inhibition of T cell proliferation, activation, cytokine production, altered metabolism and cytotoxic T lymphocytes killer functions, and eventual death of activated T cells. Clinically, antibodies targeting PD-1 or PD-L1 have been demonstrated to be able to reinvigorate the “exhausted” T cells in the TME. To demonstrate the therapeutic synergy between bispecific NIE and PD-1/PD-L1 ICB therapy, we randomized syngeneic orthotopic 4T1 breast cancer-bearing mice into four groups for anti-PD-1 Ab (anti-PD-1) therapy with or without additional nanoparticles: (1) anti-PD-1 alone; (2) regimen 4 (CNIE-NPs) plus anti-PD-1; (3) regimen 5 plus anti-PD-1; (4) regimen 6 (NIE-NPs) plus anti-PD-1. When tumour volume reached about 100 mm3, nanoparticles were given i.v. injected on day 1 (13 mg/kg), and anti-PD-1 (200 mg/mouse) given i.p. on day 2. The same cycle was repeated on day 3, 5, 7, and 9 for a total of 5 cycles, and mice were observed continuously for 21 days (Fig. 5a). Not unexpectedly, anti-PD-1 alone and regimen 4 plus anti-PD-1 treatment were ineffective (Fig. 5b). In contrast, regimen 5 plus anti-PD-1 treatment did significantly suppress tumour growth, resulting in a longer median survival, compared with 8 treatments of regimen 5 without anti-PD-1 as shown in Fig. 4b,d (49.5 d vs. 39 d); both of these treatments however were not able to completely eliminate the tumours. Most remarkably, mice treated with regimen 6 (NIE-NPs) plus anti-PD-1 resulted in gradual shrinkage and eventual complete elimination of tumours within 21 days, and without any sign of recurrence during the observation period of 90 days (Fig. 5c), validating the synergistic effects of our bispecific NIE with ICB therapy. In addition to i.v. administration, we have also investigated the therapeutic efficacy of NIE-NPs via intratumoural (i.t.) injection plus anti-PD-1 antibody given i.p. As shown in Fig. 5 and Supplementary Fig. 25, both administration routes for NIE-NPs (i.v. or i.t.) yielded excellent immunotherapeutic efficacy. However, regimen 4 (i.t.) plus anti-PD-1 and regimen 5 (i.t.) plus anti-PD-1 groups were still unable to eliminate the tumour after five cycles of treatment, while i.t. injection of regimen 6 (NIE-NPs) plus anti-PD-1 group eventually eliminated tumour and maintained 100% survival rate within 90 days.
Unlike traditional chemotherapy or targeted therapy in clinical oncology, immunotherapy can potentially induce an adaptive response with capacity for memory.40, 41 Immune memory is crucial to achieving durable tumour responses and preventing recurrence. To assess whether the synergistic therapy of bispecific NIE plus anti-PD-1 Ab could induce an immune memory response, we re-challenged the “cured” mice from previous experiment (regimen 6 plus anti-PD-1 treatment, Fig. 5a-c) with 4T1 tumour cells on the opposite mammary fat pad on day 90; naive mice of the same age were used as a negative control (Fig. 5d). The tumour volume of all the naïve mice increased rapidly within 30 days (Fig. 5e). However, either no tumour growth or significant delay in tumour growth was observed in mice previously treated successfully with NIE-NPs plus anti-PD-1 (Fig. 5f), confirming the presence of an excellent immune memory response mounted by these previously treated mice. Survival curves of this experimental group correlated well with tumour growth results (Fig. 5g). All mice remained alive during the 60-day observation period (day 90-150). In addition, the serum levels of cytokines such as TNF-a and IFN-g in this experimental group were found to be much higher than those in the control same age naïve mice group after re-challenged with 4T1 tumour cells for 6 days (Fig. 5h,i). These results suggest that a durable and robust T cell memory response was generated by regimen 6 (NIE-NPs) plus anti-PD-1 given previously.
Although the up to 40% response rate of ICB therapy in non-small cell lung cancer (NSCLC) is considered a clinical breakthrough, the overall survival of patients remains only 15% at 5 years and declines to less than 2% in patients with metastatic disease.42 We speculated that like 4T1 breast cancer model, NIE would also synergize ICB treatment of NSCLC. As expected, complete tumour regression and prolonged survival was obtained for the treatment of Lewis lung syngeneic subcutaneous murine tumour model using NIE-NPs plus anti-PD-1 (5 cycles, Fig 5j-l). No systemic toxicity and weight loss were detected.
{kind=link}