Clinical genetic analysis
The clinical phenotype of the child includes: 1 broad thumbs/toes; 2 patent foramen ovale, ASD and PDA; 3 aortic development abnormalities (i.e. aortic annulus, ascending aortic coarctation); 4 cardiac hypertrophy; 5. heart failure; 6 respiratory failure; 7 small intestinal necrosis.
There are several special phenomena worthy of attention in the patient's medical history. First, the ultrasound exam showed normal aortic development at 23 weeks of gestation, but showed that the ascending aorta and aortic arch were slightly thinner at 27-week, while the aortic annulus and aortic arch were severely constricted after birth. This change indicates that aortic dysplasia in the current case occurred in the late aortic development and deteriorated over time. Secondly, the patient's heart development is basically normal, in which case the aortic malformation is generally surgically treatable. However, after the child was born, he developed rapidly rare complications, including ventricular hypertrophy and heart failure, respiratory failure, and small intestinal necrosis. These suggest a highly complicated nature of the patient's disease, but not a simple cardiac malformation. Thirdly, that the patient's left ventricular development was normal until the child was born. In general, early aortic dysplasia in the fetus (e.g. abnormal aortic dysplasia can be detected by ultrasound at as early as 16 weeks) can lead to left ventricular dysplasia, mainly due to insufficient blood supply[10], but which did not occur in this case. The relatively small effect on left ventricular development in this case might be due to late and less serious aortic dysplasia occurred in the third trimester. Overall, this is a rare and special RTS case, whereas serious complications occurred after birth. Because of these, we decided to investigate its underlying molecular mechanisms through state-of-art technologies and approaches.
Zebrafish model
The CMA analysis of the patient showed the deletion region of 3,721,533-4,242,948 [hg19] at 16p13.3 and three genes, TRAP1, CREBBP, and ADCY9, are mapped to this region. Mutations, deletions, and duplications of the CREBBP gene cause RTS, which is typically characterized by broad thumbs and toes[11]. In animal models, no cardiac abnormalities were recorded[12]. As shown in the literature, large CREBBP deletions contribute to more severe RTS phenotypes than point mutations, implying a concurrent pathogenetic role of flanking genes, a phenomenon typically known as contiguous gene syndromes[13, 14]. Therefore, it is reasonable to hypothesize that in addition to the CREBBP gene, other genes in the same deleted region may be involved in the disease of the current patient. For the other two genes TRAP1 and ADCY9, the function of TRAP1 has been relatively clear[15, 16]. TRAP1 encodes an intra-mitochondrial protein, highly homolog to members of the Hsp90 family, which play fundamental roles in protein folding, protein degradation and signal transduction[17]. Despite these, the Trap1 knockout mice did not have heart abnormalities[18]. Therefore, we proposed ADCY9 as a plausible candidate to explain the severe heart abnormalities observed in the current patient.
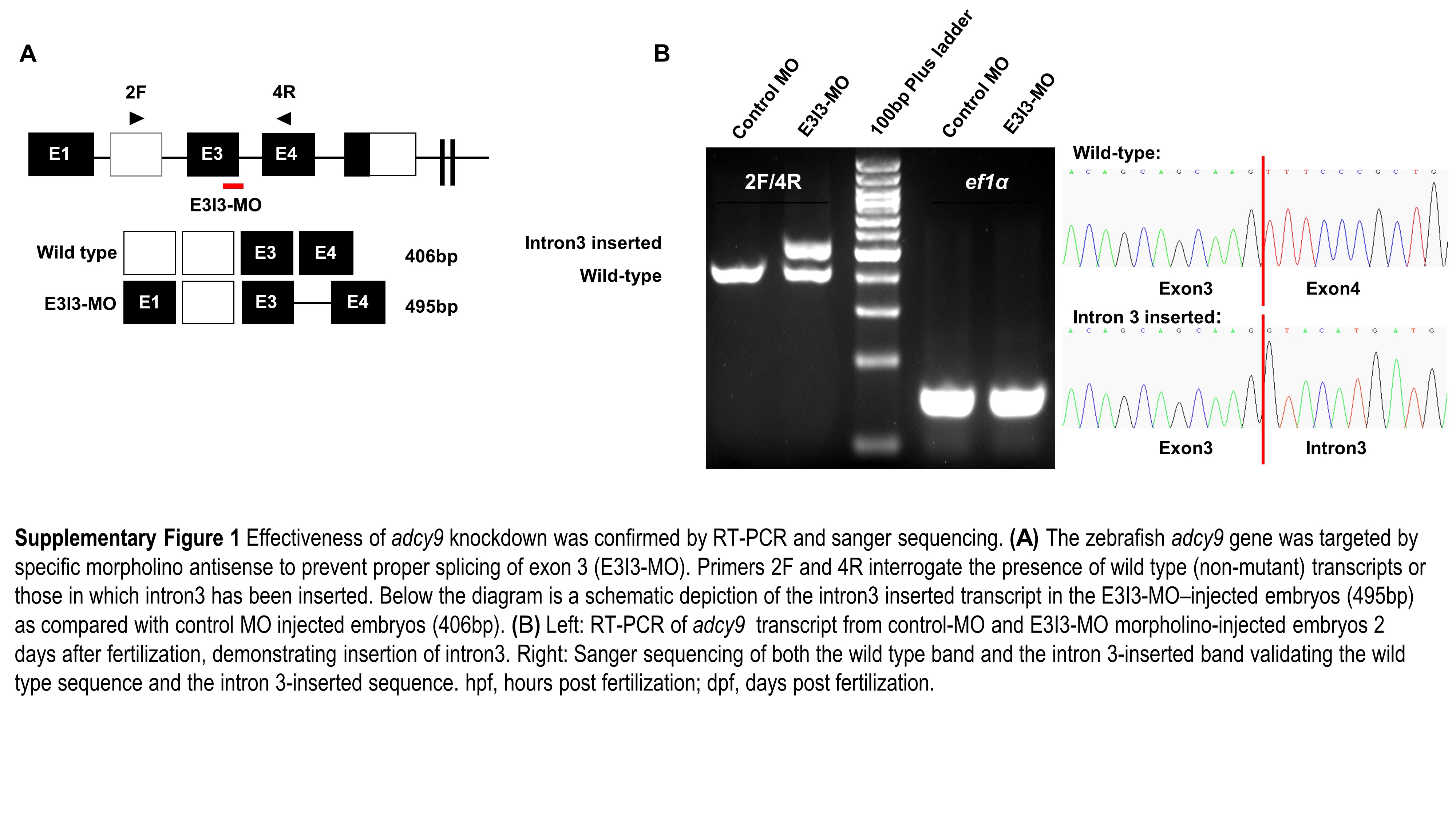
The ADCY9 gene is conserved across multiple species. Compared with zebrafish, the protein and DNA similarity rates of human ADCY9 are 65.2% and 64.7% respectively. To exam the function of ADCY9 in heart abnormalities, we designed an adcy9 knockdown experiment in zebrafish via morpholino-modified antisense oligonucleotides. RT-PCR and Sanger sequencing confirmed that the adcy9 MO experiment was successful, and the target sequence was inserted into the intron 3 of the adcy9 gene in the MO group.
Heart beat is visible and normal in the control zebrafish, but is abnormal in adcy9-e3i3-MO injected zebrafish (Fig.1, Supplementary Movie1, Supplementary Movie2). Compared with the control zebrafish, the zebrafish with the adcy9 knockdown has slower heart rate, pericardial edema, small ventricles, cardiac hypertrophy, and atrial blood stagnation. All embryos (100%) had development defects. The adcy9 morphant zebrafish has a high mortality rate of 50% at birth. The cardiac malformation of the adcy9 morphant zebrafish is mainly presented in the following two aspects: 1. the ventricle of the adcy9 morphant zebrafish is smaller than that of the control zebrafish, indicating ventricular dysplasia in the zebrafish; 2. the adcy9 morphant zebrafish generally have symptoms such as decreased heart rate, pericardial edema, cardiac hypertrophy, and atrial congestion. These indicate that the adcy9 morphant zebrafish have severe acute heart failure.
1. Immunofluorescence study on macrophage and apoptosis
The effect of adcy9 knockdown on macrophage migration was observed by immunofluorescence technique (Fig.2). Compared with control fish, embryos injected with adcy9-e3i3-MO showed increased macrophage migration, including the heart. The effect of adcy9 knockdown on zebrafish apoptosis was also observed by immunofluorescence technique. In contrast to control fish, apoptosis was specifically observed in embryonic hearts of adcy9 knockdown zebrafish (Fig.3). Real-time PCR confirmed that the apoptotic indicator in zebrafish, baxa, had increased expression, while caspases and HDR had reduced expression.
2. RNA-seq results
To investigate the molecular mechanisms of acute heart failure and high mortality of adcy9 MO zebrafish, we performed RNA sequencing analysis. The RNA-seq results of differential gene expression analysis comparing zebrafish with or without adcy9 knockdown are shown in Supplementary Table 2. Most significantly, ela2(encoding elastase 2)and cyp1a (Human ortholog gene CYP1A1, encoding a member of the cytochrome P450 superfamily of enzymes) had decreased expression in the adcy9 morphant zebrafish. In contrast, these genes had increased expression:
mmp9 (Human ortholog gene MMP9, encoding matrix metallopeptidase 9, involved in the breakdown of extracellular matrix in embryonic development);
syngap1a (Human ortholog gene SYNGAP1, encoding synaptic Ras GTPase activating protein 1);
fosl1a (Human ortholog gene FOSL1, encoding FOS like 1, AP-1 transcription factor subunit, regulating cell proliferation, differentiation, and transformation);
kank2 (Human ortholog gene KANK2, encoding KN motif and ankyrin repeat domains 2, involved in cytoskeletal formation by regulating actin polymerization);
acp5a (Human ortholog gene ACP5, encoding acid phosphatase 5, tartrate resistant);
pxnb (Human ortholog gene PXN, encoding paxillin, a cytoskeletal protein).
Limited by the sample size of the RNA-seq study, we got 17 genes with q-value<0.10, which means 10% of these genes (~2 genes) are false positive. Considering the critical roles of mmp9 in developmental growth and regeneration, we did a realtime PCR experiment and confirmed its higher expression in the morphant zebrafish.
Consequently, we performed GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses on up-regulated and down-regulated genes (Fig.4). The GO Biological Processes (BP), the GO Molecular Functions (MF), and the KEGG pathways, highlighted in this study, are shown in Supplementary Table 3. A complete list from the pathway analysis are shown in Supplementary Table 4.
{kind=link}