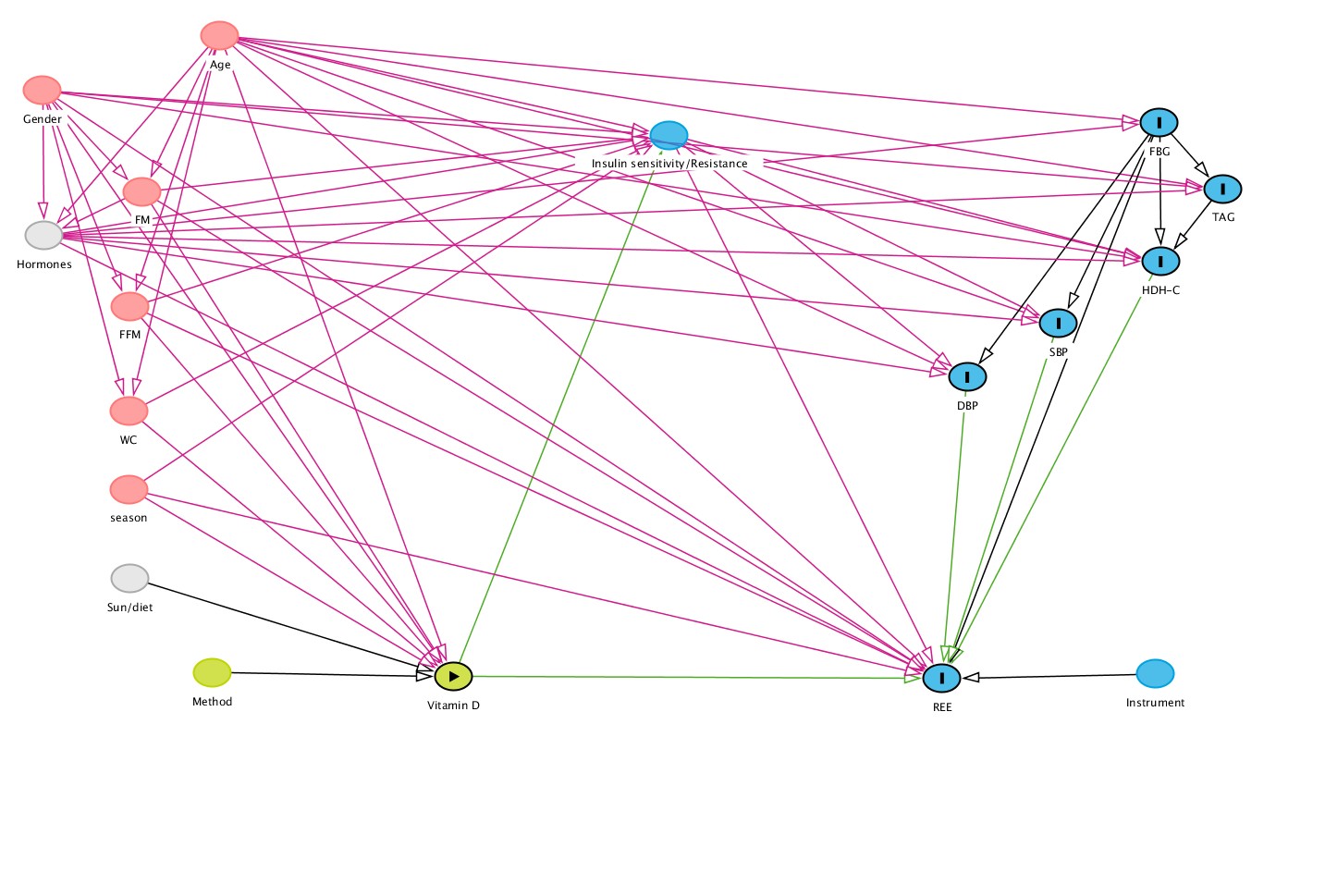
The potential role of vitamin D in the regulation of body weight in humans has been reviewed [8, 34], with animal and cellular models suggesting a role for the vitamin in energy balance. However, the precise mechanisms underpinning the influence of the vitamin on human energy expenditure are yet to be confirmed. This analysis was based on the hypothesis that insulin sensitivity (IS)/insulin resistance (IR) may mediate the association of vitamin D status (25OHD) on resting energy expenditure (REE). We observed a partial negative mediation of IS/IR on the positive relationship between higher vitamin D status and increased REE. The magnitude of this mediation indicated that any projected increases in REE following vitamin D supplementation, would be significantly dampened by concomitant improvements in IS or reductions in IR (Table 2).
1,25-dihydroxyvitamin D (1,25(OH)2D) is the major active form of the vitamin, and exerts its effects via the vitamin D receptor (VDR). Global VDR null mice, when compared to wild type mice, displayed a lower body weight even on a high fat diet [6]. This arose from a greater energy expenditure and an upregulation of uncoupling proteins (UCP) in adipose tissue. Another pathway for increased energy expenditure in VDR null mice could involve the higher bile acid pool seen in these animals [34]. Interestingly, targeted over expression of VDR in adipose tissue of mice, led to a higher body weight and specifically greater fat mass [7]. There was no change in food intake in these animals, but energy expenditure was reduced, partly from a suppression of UCP [7]. Further, cellular models of research indicate that treatment with 1,25(OH)2D suppressed UCP expression in primary cultures of brown adipose tissue (BAT) [6, 7]. Extrapolating outcomes from animal models to humans is not straightforward, and the discrepancies are not easily understood [34]. Currently, human studies that have addressed this area are few in number, and present their own issues. For example, a cross sectional study from Iran reported that 25OHD was positively related to REE/kg body weight [22]. Besides the manner of expression of REE, those authors further adjusted REE/kg weight for fat free mass and other covariates, which makes interpretation difficult. Another cross sectional study from the same country, where ~ 80% of the participants had 25OHD < 30 nmol/L, reported no relationship between 25OHD and REE adjusted for several covariates including a measure of IR [35]. There are three supplementation studies to date in the literature. A 1-week intervention found no effect on energy metabolism or substrate utilization, following vitamin D supplementation [36]. A longer 6-month trial was conducted on individuals with vitamin D deficiency and type 2 diabetes [37], where the supplementation followed a bolus dose regimen. The vitamin D supplemented group showed a small but significant decrease in absolute REE, but that data was not adjusted for any change in body composition over the trial period [37]. In addition, the authors reported that insulin sensitivity, as measured by the gold standard hyperinsulinemic-euglycemic clamp technique, was not different [37]. An acknowledged limitation of that study was a steady decrease in 25OHD over time. By trial completion at 6 months, the final status had a mean (SD) of 54(9.2) nmol/l. There is a growing view that a threshold level of 25OHD > 75 nmol/L, is required for non-skeletal effects of the vitamin [11, 38, 39]. Vitamin D replete individuals (baseline 94 nmol/L) assigned to a treatment group, increased their 25OHD by ~ 30 nmol/L but showed no difference in their raised REE compared to the placebo group (baseline ~ 74 nmol/L) who showed a decline in their status. [40]. Indirect evidence in humans comes from the close relationship of mitochondrial oxygen affinity with REE [41], and that mitochondrial oxidative function of skeletal muscle improved with vitamin D supplementation [42]. Overall, consistent direct human evidence in support or against the hypothesis is lacking, and so future research is required to test the paradigm generated here.
Previous RCTS investigating the impact of vitamin D on IS/IR had been critiqued for their methodological flaws and other limitations. Mostly, these RCTS were not primarily designed for evaluating glycaemic-related outcomes, and they used inappropriate or infrequent doses that did not sustain vitamin D concentration. A recent meta-analysis that addressed these concerns was based on 28 RCTs identified through a systematic review. The author’s showed a reduction in IR following vitamin D supplementation [43]. There are several potential mechanisms, that may underscore this outcome, and its subsequent impact on energy metabolism. Vitamin D may increase insulin secretion via modulation of calcium concentration in pancreatic β cells [17, 44]. Vitamin D could also directly decrease pro-inflammatory cytokines, which are associated with IR [45]. Cytokines act through plasma cell membrane receptors to increase energy expenditure [46], so any decrease in pro-inflammatory cytokines, would reduce energy expenditure. The older literature in support comes from observations that basal endogenous glucose output, fasting insulin, free fatty acid concentrations, and glucose disposal were all significant determinants of REE [47]. An observational human study also found a positive relationship between changes in fasting glycemia, and changes in REE, 24h energy expenditure and sleeping metabolic rate respectively; where greater hyperglycemia promoted higher rates of energy expenditure and a reduced risk of weight gain [48].
In this mediation analysis, we found consistent evidence for a sizeable direct pathway between 25OHD and REE, however only 2 of 3 adjusted models showed a statistically significant effect (Table 3). There was also a significant, negative mediation pathway through both McA and TGY, but not with QUICKI. Those negative mediation effects blunted the overall effect across all markers of IS/IR (Table 3). The absence of a significant mediation effect with QUICKI was unexpected. All three IS/IR variables were significantly related to each other, and to REE and 25OHD respectively, in a simple correlation analysis (Table S1). Clearly the mediation effect, as a proportion of the direct effect, was ~ 50% lower for QUICKI in the fully adjusted model, in comparison to the other two surrogate measures (Table 3). So, we clearly lacked the power to detect this unexpectedly smaller effect. However it is also likely that all surrogate markers of IS/IR are not equally efficient in predicting downstream changes in energy expenditure. Other authors working on different clinical endpoints have reached similar conclusions [49–51]. They too noted significant simple correlations between all markers and their study endpoints (e.g. metabolic factors [49], cardiovascular disease [50] or type 2 diabetes [51]), but with fully adjusted models the predictive ability of each surrogate marker was quite different. Perhaps, the way forward in this area would be the inclusion of several surrogate markers, especially when studying other global populations/ethnic groups.
Limitations
These cross sectional data, restricted to one ethnic group, are not representative of the present Australian population nor of other populations groups worldwide. Some unexpectedly small effect sizes were not detected, with resultant p values < 10%. While this is acceptable for hypothesis generation, a much larger cohort would have overcome this limitation. Moreover, a larger sample would have allowed the modelling to be developed on one random half, and validated on the other half within this paper. We have identified some unmeasured variables, mainly circulating hormones like thyroid, parathyroid hormone, leptin, cortisol, adiponectin etc. that could make small contributions to residual variation in REE, and impinge on IS/IR as well. Their future inclusion could be important to clarifying the precise contributions of 25OHD and IS to REE.
{kind=link}
{kind=link}