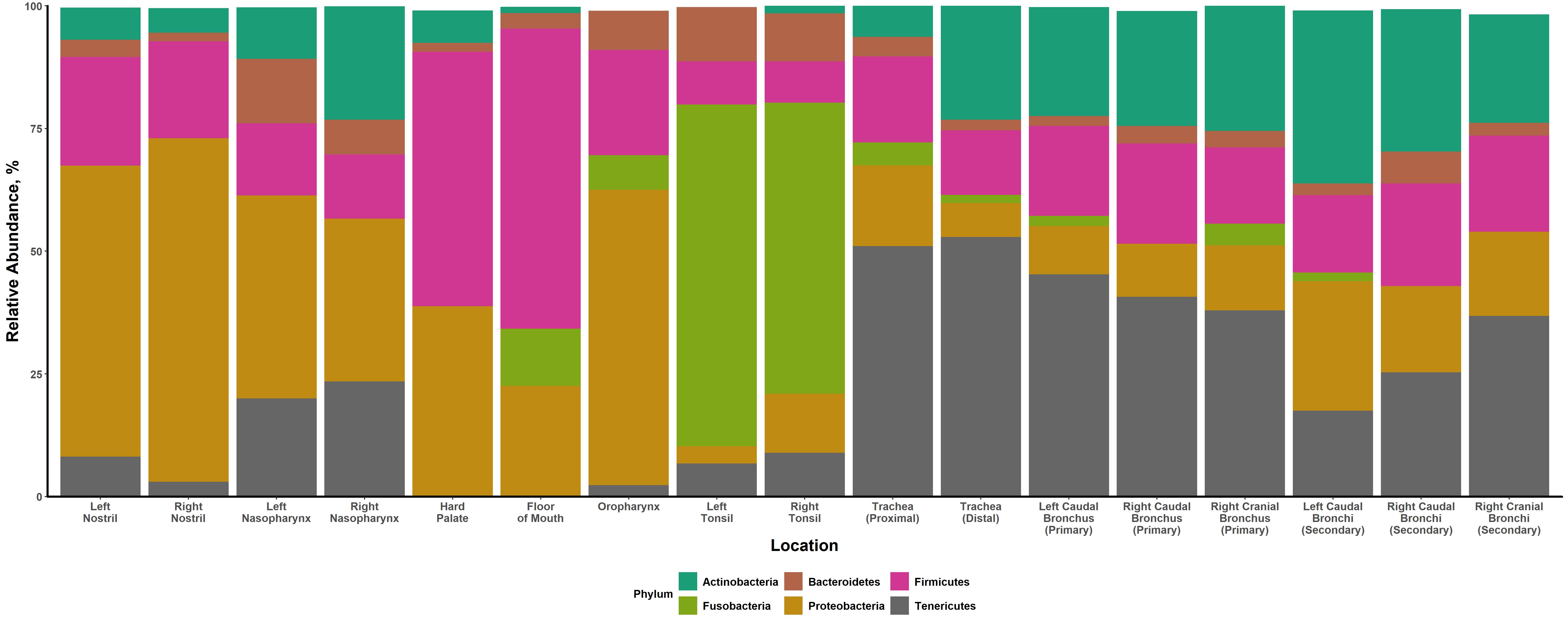
This study was the first to describe and compare the bacterial microbiota of different niches present along the entire respiratory tract of healthy cattle and determined which URT microbiota are most similar to the composition of the lung microbiota. We showed that community composition and diversity differed among different niche microbiota. These differences were driven by a variety of taxa, notably Mycoplasma, Moraxella, Streptococcus, and Fusobacterium. We also showed that the lung microbiota was more compositionally similar to the nasopharynx than any other URT microbiota, including the nostrils, oropharynx, and tonsils. This finding indicates that the nasopharynx is likely the primary source of bacteria for the lung in healthy cattle.
The characterization of distinct niche microbiota throughout the bovine respiratory tract is a novel finding. Compositional differences between URT and lower respiratory tract (LRT) microbiotas have been previously observed in cattle [13–15]. However, these past studies have been limited in scope, comparing only trans-tracheal aspirations or bronchoalveolar lavages to the nasopharynx. The findings of the current study corroborate what has been seen in humans and other ruminants. Previous research has shown humans to have marked dissimilarity between the nasal, oral, and lung microbiotas [7, 16, 17]. As well, the microbiota of the oral cavity in lambs has been reported to be distinct from the lung [18].
The presence of distinct niche microbiota across the respiratory tract was expected as there are known physiological and biochemical differences among the many different respiratory locations. Spatial heterogeneity in pH, CO2 levels, temperature, epithelial cell types, mucosae thickness, and immune cells have been found throughout the respiratory tract, with human studies even showing heterogeneity in different regions of the lungs [17, 19–22]. The URT is also under constant external pressure from the surrounding environment, which can significantly impact community composition [22].
Despite the characterization of numerous distinct microbiotas throughout the respiratory tract, noticeable compositional overlap was still observed between various niches. Interestingly, though there was significant variation in bacterial composition between sampling locations, there was limited variation across lung sites or between anatomically similar sampling locations (i.e. left and right nostrils, left and right nasopharynx, left and right tonsils). Research in humans has shown that there is little spatial variation across different lung sites within healthy individuals [17]. Yet, in sheep it was found that, depending on the individual animal, there may or may not be significant variation between lung sites [21].
The lack of bacterial variation that we observed within the lung may be partially explained by the adapted island model of lung microbiota biogeography [19]. In this model, the bacterial composition of the lung is determined more by the constant flow of transient bacteria than the replication of resident bacteria [17, 23]. Foreign bacteria are constantly migrating into the respiratory tract through a combination of inhalation, aerosolized saliva microaspiration, and dispersion along mucosal surfaces [19, 24–26]. At the same time, these bacteria are also being constantly cleared from the respiratory tract by forced exhalation (coughing/sneezing) and host respiratory defenses (i.e. mucociliary clearance, antimicrobial peptides, immunoglobulin A, immune defense cells, etc.) [26, 27]. This model concept supports our finding that compositional overlap between URT and LRT microbiotas occurred, and that the URT microbiotas were a source of bacteria residing in the lungs of healthy cattle. It should be noted that this model may not apply to sick animals, as it has been shown that the progression of lung diseases (such as cystic fibrosis) can impair bacterial clearance, leading to increased colonization and proliferation of bacteria in the lung [28, 29]. Regardless, understanding which URT microbiotas contribute the most to the healthy lung is a key component in understanding the complexities of the respiratory system in cattle.
Contrary to what has been seen in humans, the oral microbiotas (specifically the oropharynx) have less compositional overlap with the lung than the nasopharynx. It has been proposed that, in healthy adult humans, aerosolized saliva containing bacteria are aspirated into the lungs during sleep as a result of the throat muscles relaxing [7, 23]. While the oropharynx has been suggested as the primary source of bacteria to the lung in healthy adult humans, it has also been suggested that the oropharynx and nasopharynx both contribute to the lung microbiota in healthy neonates [30]. This process may not be the same in cattle though, as ruminants have notable differences in respiratory anatomy and physiology compared to humans. A study performed in lambs compared the oropharyngeal and ruminal microbiotas with the lung and found that they were significantly dissimilar [18]. These differences might be explained by the horizontal disposition of the lung or by evolutionary anatomical barriers to microaspiration of ruminal fluid into the lung [18]. It is also worth noting that the bovine trachea is longer relative to body size compared to other animals, which may also add to the differences observed between human and ruminant respiratory microbial compositions [27].
That the nasopharyngeal microbiota is the primary source of bacteria for the lung has practical implications. Notably, the nasopharynx (but not the nostrils) appears to be the most important location we should be sampling when doing microbiota research to better understand BP. As well, the nasopharynx is likely the optimal microbiota to target for novel therapeutics such as probiotics, phages, etc., to mitigate BP pathogens.
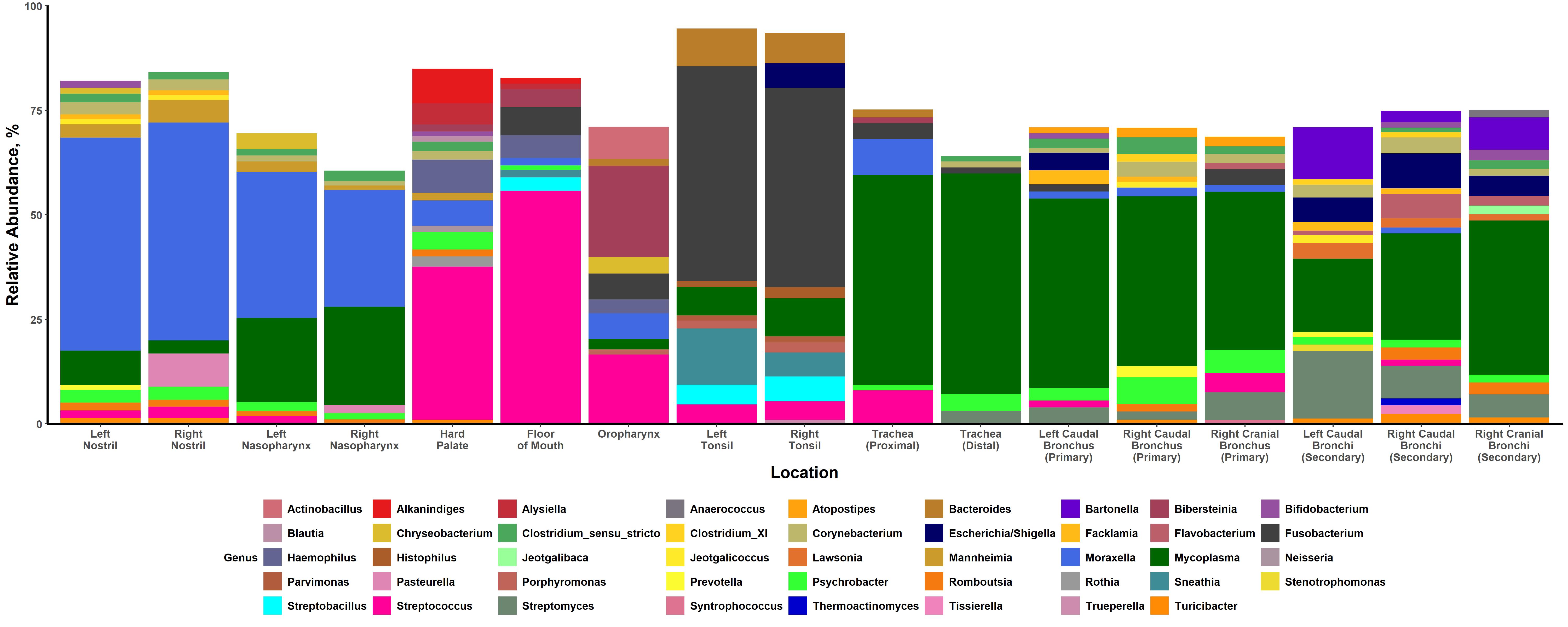
It is possible that the oral microbiotas in cattle act as secondary bacterial reservoirs for the lung. The oral microbiotas were primarily dominated by the genus Streptococcus in the current study, corroborating what has been previously seen [31, 32]. Interestingly, we were able to identify a metacommunity dominated by Streptococcus in the oral microbiotas as well as the lung, though we were unable to identify most of the bacteria in this genus at the species level. Greater clarity at the species level could be valuable, as overlap between the ruminal and oral microbiotas has been previously observed [33]. The process of rumination, whereby cattle regurgitate feed to be masticated a second time before swallowing the feed again, is integral to the digestion in cattle [33]. Streptococcus bovis, a known rumen inhabitant that plays an increased digestive role in cattle that are being transitioned to high starch diets, has the potential to gain entry to the oral cavity via rumination [33–35]. In the current study, cattle were in the process of transitioning to a high starch diet (i.e. corn-based) at the time of sampling, providing a potential explanation for the observed abundance of Streptococcus in the oral microbiotas. Whether the Streptococcus observed in the oral and lung microbiotas in this study are S. bovis or a different Streptococcal species is unclear, and further research is needed to understand the relationships between the ruminal, oral, and lung microbiotas.
The palatine tonsils may also act as a secondary reservoir of bacteria for the lung microbiotas. We observed that the tonsils were dominated by the bacterium F. necrophorum in the current study. This finding is notable, as F. necrophorum is a normal inhabitant of the rumen and also the primary causative pathogen for liver abscesses in cattle [36]. We were able to identify a metacommunity dominated by F. necrophorum in both the tonsillar microbiotas and the lung. This again suggests not only a possible link between these microbiotas, but also between the rumen and the lung microbiotas. Additionally, we found at least one of H. somni, P. multocida, or Mycoplasma bovis in the tonsils of each calf, with multiple calves harboring more than one of these pathogens. In contrast, M. haemolytica was only found in the tonsils of one calf. Previous research has shown that tonsils inoculated with M. haemolytica can serve as a reservoir for the bacterium [8, 37–39]. During periods of stress or respiratory viral infection M. haemolytica can be shed from the tonsils [37–39]. However, in healthy calves it has been shown that M. haemolytica shed from the tonsils into the nasal mucous are rapidly cleared from the nasal passages [8, 38, 39]. As we only studied healthy animals, we cannot rule out the tonsillar microbiota as a potential source of bacterial pathogens for the lung.
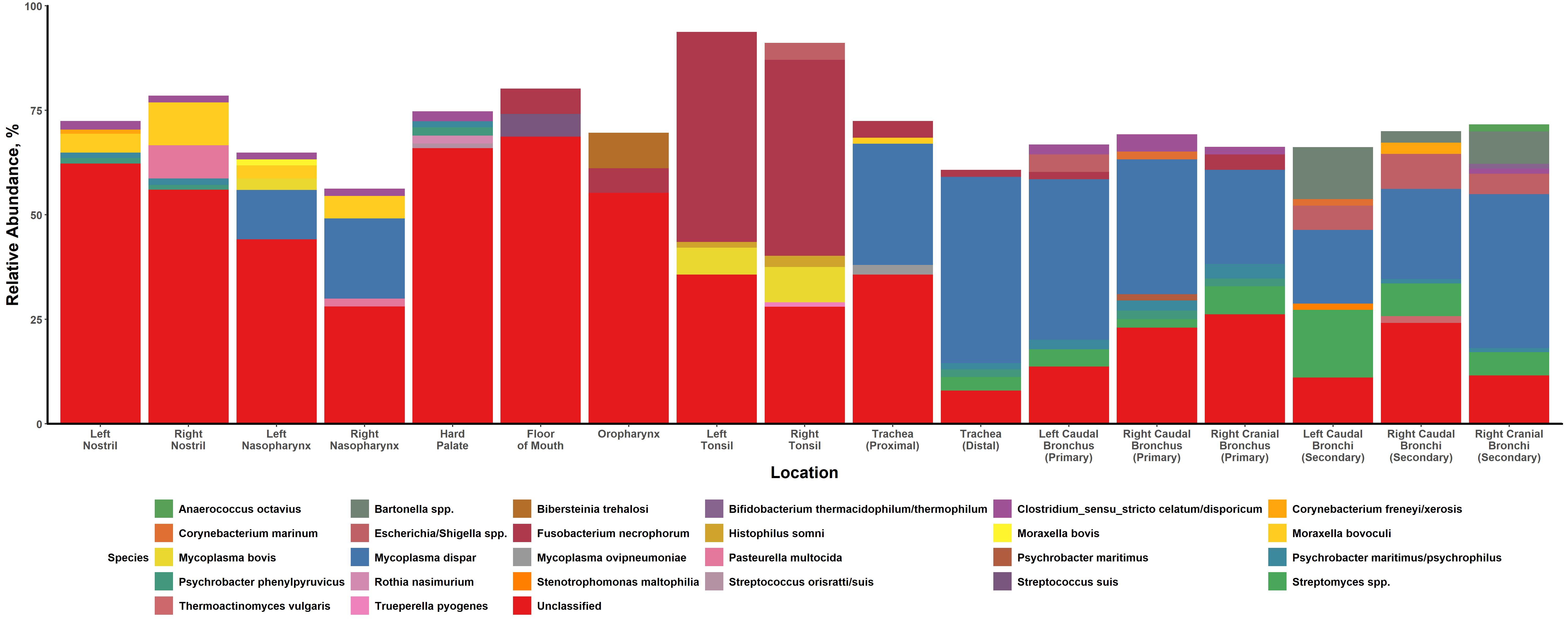
The high abundance of Moraxella in the nasal passageways observed in the current study agreed with findings from previous research. Moraxella is often found to be one of the most abundant genera in the URT of cattle [14, 40–42]. The role of this genus in BP has been brought into question before, with one previous study finding an association between Moraxella and the development of pneumonia and/or otitis in early life of dairy calves [43]. It is also known that M. bovoculi and Moraxella bovis are opportunistic pathogens, commonly accepted as being the primary etiological agents of infectious bovine keratoconjunctivitis (IBK) [44, 45]. In contrast, another study found that M. bovoculi was one of the top bacterial species driving differences in community composition between healthy cattle and those that had developed BP, with a higher abundance of the bacterium found in the nasopharynx of healthy calves [40]. Thus, the strain of Moraxella may determine pathogenicity and subsequent roles, if any, in BP. Indeed, it has been shown that M. bovoculi isolated from the eyes of cattle that had developed IBK were significantly different at a genomic level from nasopharyngeal isolates found in healthy cattle [46]. That we found a high abundance of Moraxella, more specifically M. bovoculi, in both the nostrils and nasopharynx of healthy cattle might suggest that certain strains of these bacteria may be part of the normal, healthy nasal microbiota. Future research using whole genome sequencing techniques could provide greater clarity at lower levels of taxonomic identification on what role Moraxella may play in cattle respiratory health.
Mycoplasma, specifically M. dispar, was frequently identified in both the lung and nasopharynx, with the highest relative abundances observed in the lung. This finding echoes a number of other studies that have reported Mycoplasma as one of the most commonly identified genera in the nasopharynx and lung [13, 14, 40–42, 47]. It is not clear what role M. dispar plays in respiratory health. This bacterium has been previously isolated from the lungs of both healthy and pneumonic cattle [48–50]. Interestingly, a study by Timsit et al., 2018 identified a distinct metacommunity which was characterized by an over-representation of M. dispar (and other commensal bacteria such as Lactococcus lactis and Lactobacillus casei) in the lungs of healthy feedlot cattle. While M. dispar has been shown to have a number of virulence factors for bovine epithelial cells and can have immunosuppressive effects, it is associated with only milder respiratory infections and is likely not a causative agent for BP [48, 50, 51]. In general, Mycoplasma have a high affinity for binding to respiratory epithelial cells via adhesin proteins [52, 53]. That M. dispar elicits a milder cellular response might explain why we found this bacterium in such high abundance in the lungs of healthy calves compared to more pathogenic Mycoplasma. A previous study that compared two genetically similar Mycoplasma species, Mycoplasma hyopneumoniae (a causative agent of porcine enzootic pneumonia) and Mycoplasma flocculare (a regular commensal bacterium in the respiratory tract of swine), found that differences in orthologous surface proteins were associated with distinct immunological responses, potentially affecting bacterial survivability [54]. Whether M. dispar confers some form of protective effect to the host (such as inhibiting Mycoplasma bovis colonization in healthy cattle by competing for adhesion sites) or is simply a common respiratory commensal bacterium remains to be determined. It would be valuable to understand when and how the nasopharynx and lungs of healthy calves are colonized by M. dispar, as previous studies have reported increased abundance/isolation of the bacterium over the first half-year of life and notably after weaning [49, 55].
{kind=link}
{kind=link}
{kind=link}