The emergence of drug resistance and limited availability of inhibitors targeting multiple cell cycle modulators pose challenges that impede the ability to halt the uncontrolled growth of cancer, leading to malignancy. Although there are currently no clinically available inhibitors or drugs for CCT, the complex and its subunits have garnered increased attention in the field of drug development54, 55. Our study revealed that, among the nine CCT subunits, CCT6A showed the highest level of activation in colon cancer. The expression of CCT6A was significantly upregulated in multiple colon cancer cell lines and clinical samples, resulting in a decreased survival rate among patients. Our findings suggest that CCT6A plays a crucial role in regulating key cellular signaling pathways involved in cell proliferation and programmed cell death, including the cell cycle, p53 signaling pathway, and apoptosis. Moreover, the inhibition of CCT6A was shown to effectively reduce tumor cell proliferation both in vivo and in vitro. These results highlight the potential of targeting CCT6A, a novel oncogene associated with colon cancer, as a therapeutic intervention.
Mutations in the TP53 tumor suppressor gene are highly prevalent in cancer; however, efforts to restore p53 functionality as a therapeutic strategy had limited success in late-stage clinical trials, with no approved p53-based therapeutics in the USA or Europe21, 56. This is because most drugs work by inhibiting excessive protein activity. Consequently, p53 has long been considered undruggable56. Although several small molecules, such as PRIMA-1, MIRA-1, and STIMA-1, have been developed to specifically target Mutp53, none of them have progressed to clinical trials owing to challenges related to solubility and toxicity towards normal cells56–58. Therefore, CCT6A may provide a novel approach for targeting Mutp53.
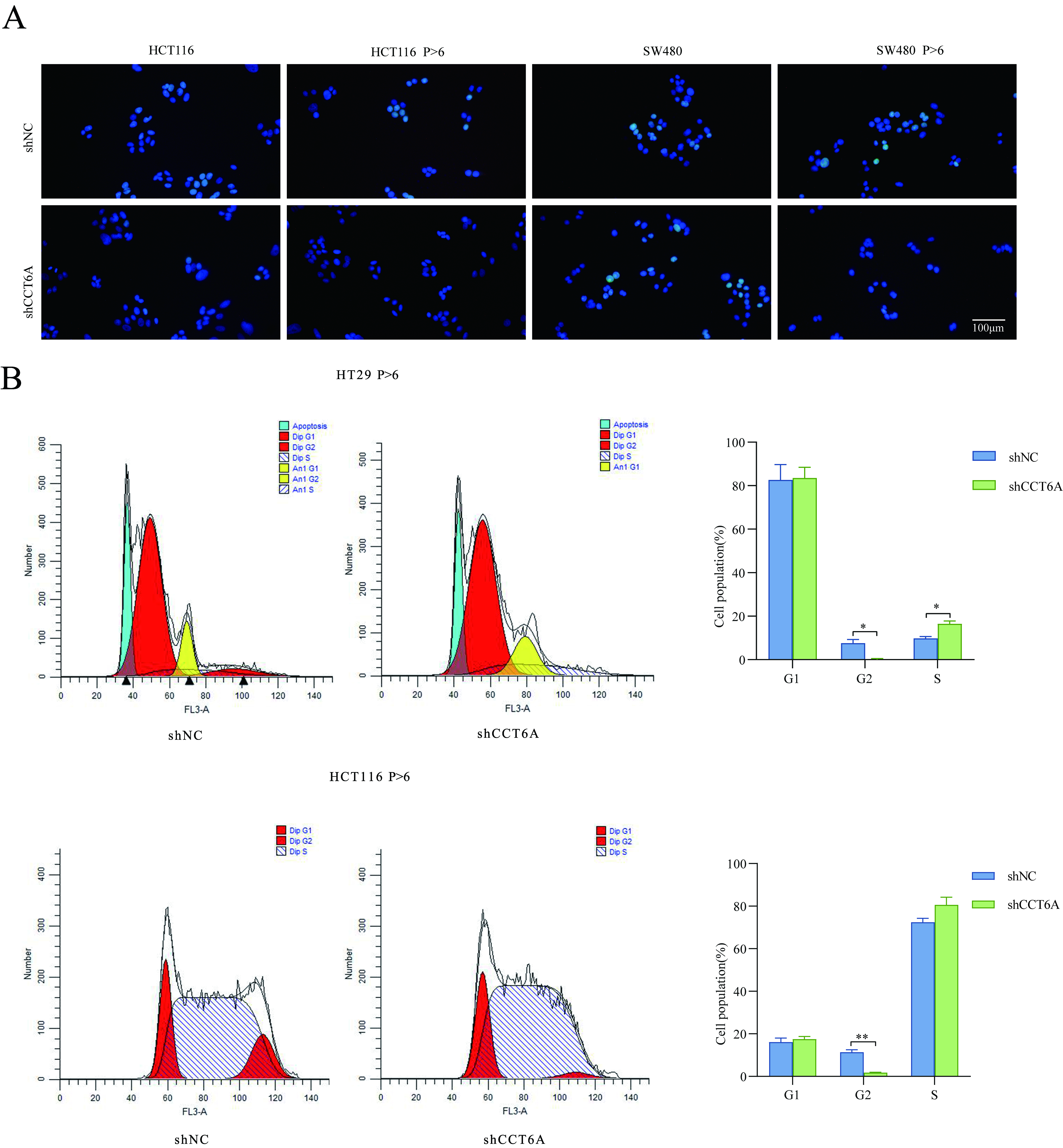
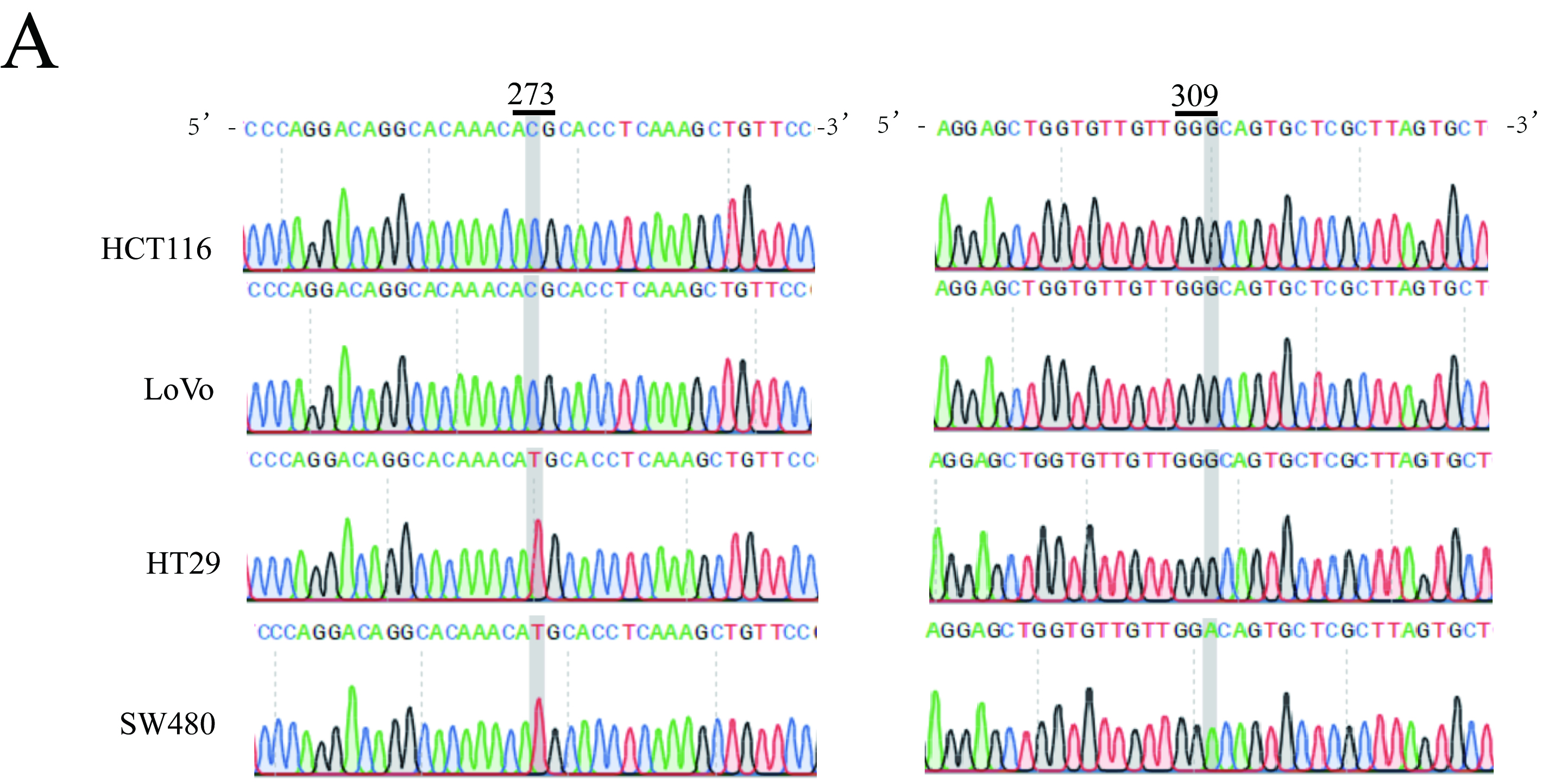
Inhibition of CCT6A expression effectively suppressed cell proliferation in both Wtp53 and Mutp53 cell lines. Interestingly, in Mutp53 colon cancer cell lines, the downregulation of CCT6A expression resulted in a reduction in the protein levels of p53, whereas the overexpression of CCT6A impeded the degradation of p53. However, this phenomenon was not observed in the Wtp53 colon cancer cell lines. Therefore, it can be concluded that CCT6A exhibits distinct functions in Wtp53 and Mutp53 cell lines. Further investigation revealed direct binding between CCT6A and both Wtp53 and Mutp53. Most cancer-associated mutations in p53 can be classified as DNA contact mutants, which exhibit only mild alterations in the overall protein structure, and conformational mutants, in which the protein undergoes extensive misfolding59–61. Previous studies established that CCT6A, serving as a constituent subunit of CCT, plays a crucial role in facilitating protein folding38. Based on these studies, we hypothesized that Mutp53 undergoes rapid folding after translation, owing to the presence of highly expressed CCT6A and other subunits of CCT. We found that reducing the protein levels of CCT6A caused the folding rate of Mutp53 to decelerate, and partially misfolded p53 was identified and modified, ultimately resulting in protein degradation; however, this process was time-consuming. This hypothesis could explain the results of subsequent nude mouse transplantation tumor experiments. However, the molecular mechanisms underlying the interaction between CCT6A and p53 as well as the degradation of p53 by CCT6A remain unknown.
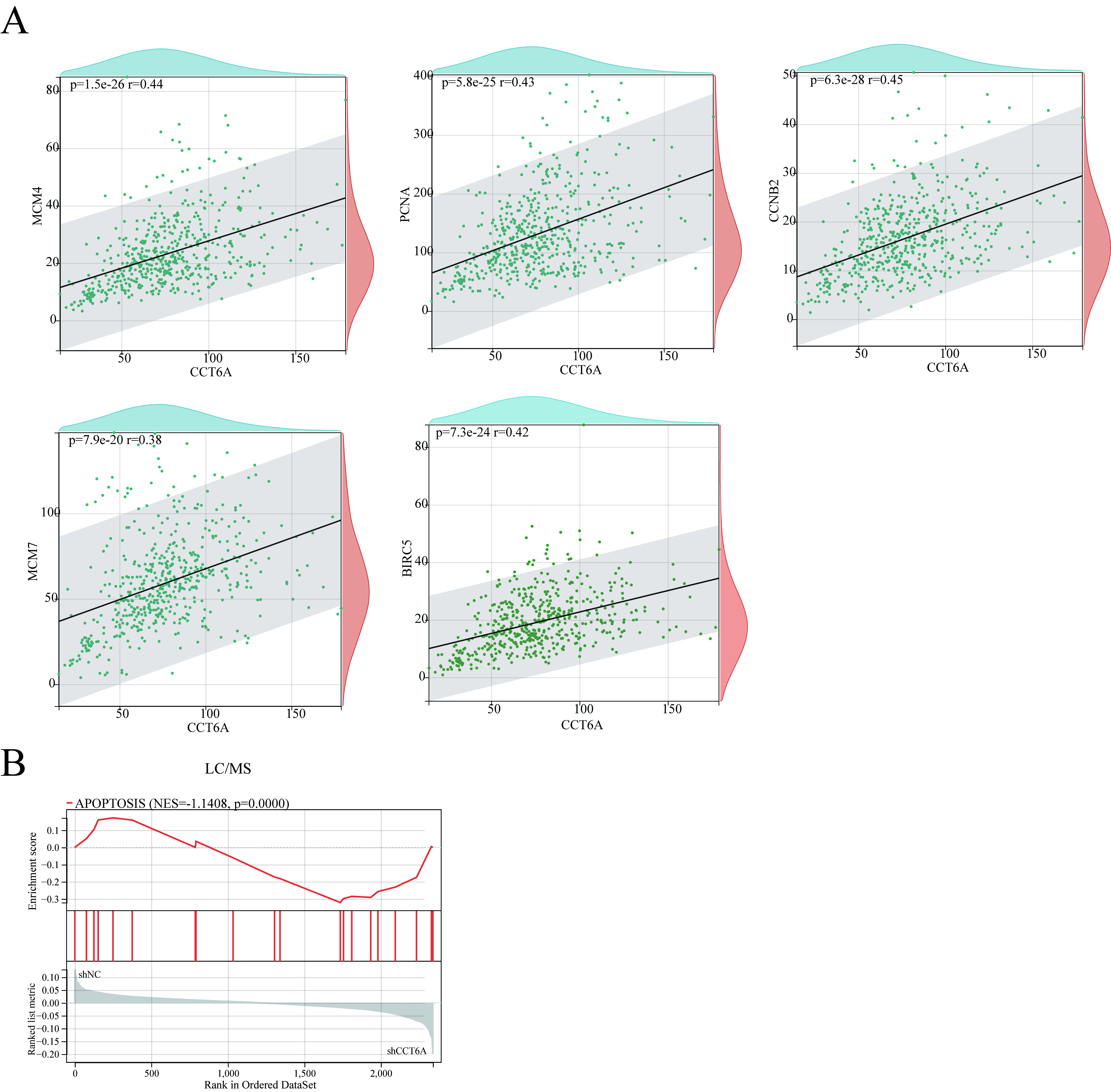
In this study, we identified the involvement of CCT6A in the regulation of BIRC5 expression. Consistent with previous studies, inhibition of CCT6A in Wtp53 colon cancer cell lines significantly reduced BIRC5 expression, regardless of p53 levels. However, the effect of CCT6A inhibition was dependent on Mutp53 levels in Mutp53 cell lines. We conclude that BIRC5 is downstream of CCT6A, but CCT6A may regulate BIRC5 through alternative pathways in Wtp53 and Mutp53 cell lines, necessitating further investigation into the underlying molecular mechanisms.
A previous study showed that BIRC5 exhibits multiple splice variants, each possessing distinct functions that confer a multifunctional nature to the protein in various cellular contexts31. The majority of these variants exert an anti-apoptotic role, such as BIRC5-ΔEx3, BIRC5-2B, BIRC5-3B, and BIRC5-3α31, 62, 63; however, BIRC5-2α demonstrates apoptotic functionality31. Our results demonstrated that CCT6A regulates the full-length BIRC5 protein and the BIRC5-3α and BIRC5-3B isoforms. BIRC5 is thought to control cell division, with cytoplasmic/mitochondrial BIRC5 displaying a protective function52 and nuclear BIRC5 being a prognostic factor for patient survival64. We found that decreasing CCT6A did not inhibit the nuclear translocation of full-length BIRC5 and its 3α and 3B isoforms. Overexpression of these three BIRC5 forms could restore the suppression of cellular proliferation caused by CCT6A silencing. Given the significance of BIRC5 as an oncotherapeutic target, its multiple isoforms warrant further investigation.
In summary, the present study highlighted CCT6A as a potential oncogene implicated in cancer progression. Importantly, CCT6A exerts its influence on BIRC5 through distinct pathways in both Wtp53 and Mutp53 cell lines. This study provides novel insights into the functional roles of CCT6A, and identifies it as a promising therapeutic target for prognosis and treatment of colon cancer.
{kind=link}
{kind=link}
{kind=link}