To date, the conventional chemotherapy treatments in patients with HCC are still not ideal due to the frequently occurring chemoresistance(21, 22). A growing number of studies have showed that the exiting of cancer stem cells and their enrichment during chemotherapy is closely associated with chemotherapy failure and HCC relapse(23–25). Thus an effective strategy to sensitize HCC to chemotherapeutic agents was to inhibit HCC stemness properties.
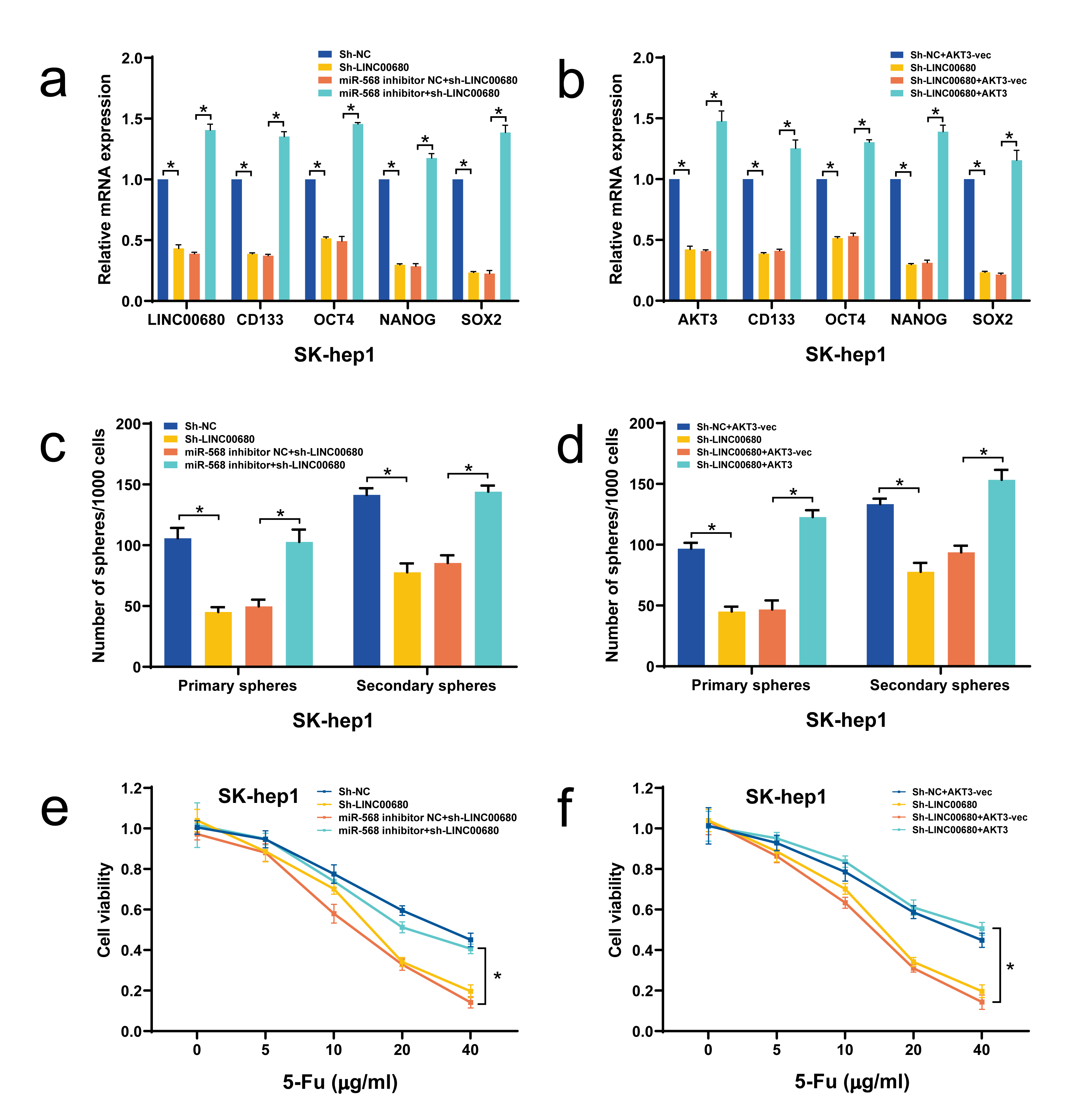
An increasing number of studies have demonstrated the critical modulating role of lncRNAs in tumor stemness phenotype as well as chemosensitivity(26, 27). In the current study, we for the first time systematically explored the potential significance of LINC00680 in HCC stemness properties and sensitivity to conventional HCC chemotherapeutic drugs, such as 5-Fu. Our data unveiled a significantly upregulated expression of LINC00680 in HCC tissues and cell lines, when compared with the adjacent normal liver tissues and normal liver cell line LO2, respectively. LINC00680 is a 1989 bp long intergenic lncRNA. So far, the functional significance of LINC00680 in HCC is completely unknown. Actually, currently there exists only one study reporting its oncogenic role in glioma(12). In our study, it was found that LINC00680 was positively associated with HCC tumor size and stage, vascular invasion, and poor survival prognosis, suggesting that LINC00680 might play an important role in HCC malignancy. Of note, our subsequent data confirmed that LINC00680 can significantly enhanced HCC stemness, as evidenced by increased primary and secondary sphere formation and HCC markers. In addition, LINC00680 also decrease HCC chemosensitivity to 5-Fu both in vitro and in vivo.
Based on the above findings, we further analyzed the underlying molecular mechanism by which LINC00680 modulated HCC stemness and chemosensitivity. Mounting information supports that sequestering miRNA to regulate mRNA expression via acting as a competing endogenous RNA (ceRNA) represents a commonly used mechanism by lncRNAs to affect HCC progression(28, 29). Bioinformatics analysis suggests that LINC00680 has putative binding sites for miR-568, which was further confirmed by our luciferase reporter, RIP, and RNA pull down assays. In addition, manipulation of LINC00680 led to a correspondingly reverse alteration of miR-568 expression. Moreover, a significant downregulation of miR-568 was seen in HCC tumor tissues. All these data imply that LINC00680 is very likely to exert its biological function via sponging miR-568.
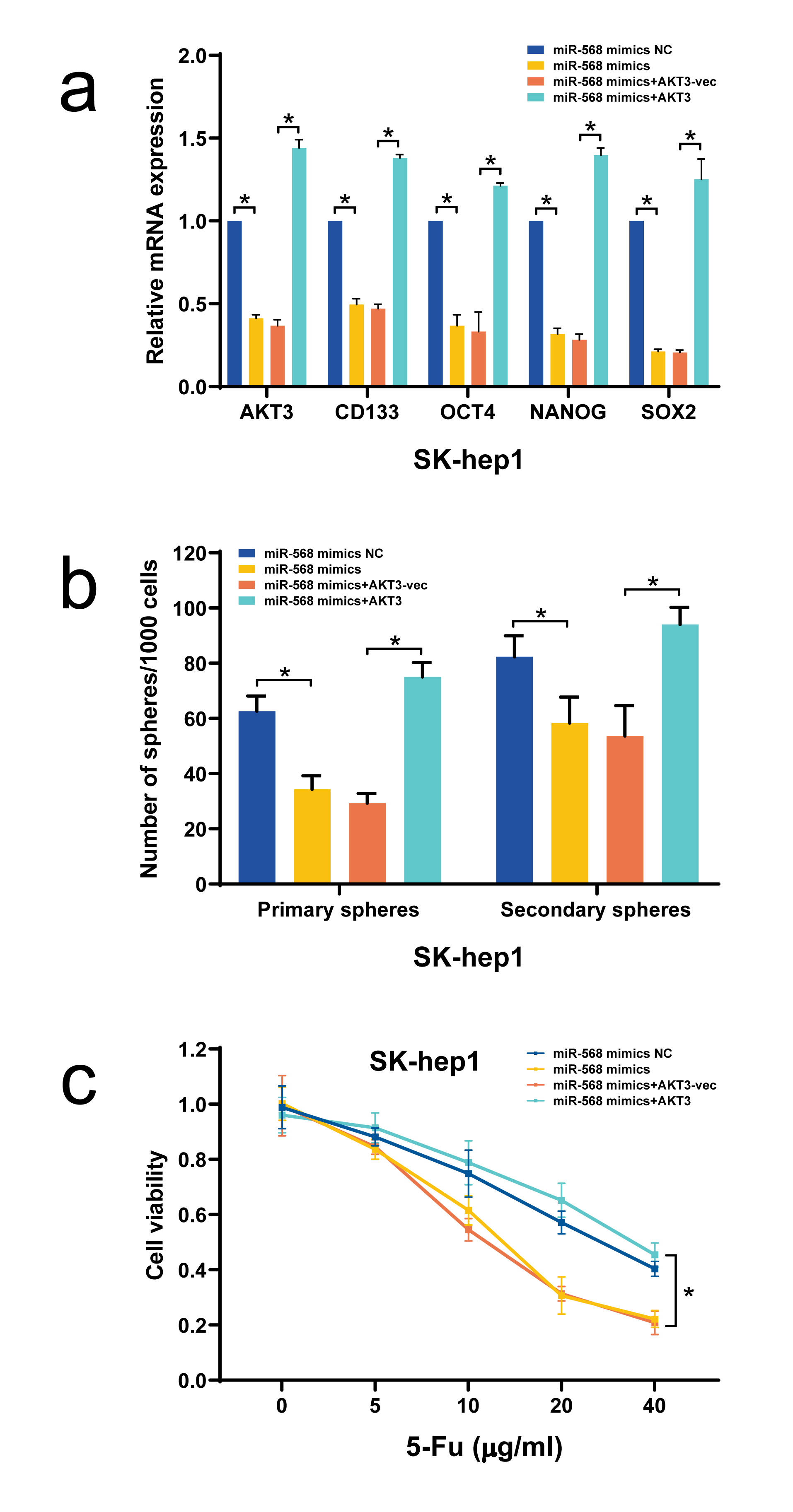
In view of that the biological functions of miRNA-568 in tumor diseases, including HCC, are completely unknown, we then determined whether miRNA-568 per se affected HCC progression and found that miRNA-568 could significantly weakened HCC stemness and correspondingly enhanced chemosensitivity to 5-Fu. Using microarray chip assay, it was found that PI3K-AKT signaling pathway was remarkably affected upon miR-568 overexpression, coupled with downregulation of AKT3. Given the well-recognized tumor-promoting role of AKT3 as reported previously(15, 30, 31), we thus reckoned that miR-568 might function as an HCC suppressor in HCC via targeting degradation of AKT3. This hypothesis was strongly supported by our experimental results, which, deriving from luciferase reporter assay, loss- and gain-of-function experiments, and expression correlation analysis in HCC tissues, demonstrated the miR-568 indeed could direct influence AKT3 level in HCC cells. Moreover, we also found that miR-568 weakened HCC stemness and enhanced chemosensitivity to 5-Fu were effectively reversed by AKT3 overexpression.
The mammalian target of rapamycin (mTOR) is aberrantly phosphorylated and activated by PI3K-AKT signaling in many human malignancies, including HCC, and plays a critical role in HCC oncogenesis(18, 32, 33). mTOR is a serine/threonine protein kinase and exist as two distinct forms: mTORC1 and mTORC2(34, 35). Rapamycin-sensitive mTORC1 promote HCC growth and progression via directly targeting phosphorylation and activation of ribosomal protein S6 kinase (p70S6K) and eukaryotic translation initiation factor 4E-binding protein 1 (eIF4EBP1)(36). Moreover, Inhibition of mTORC1 by rapamycin has been reported to sensitive HCC cells to a variety of chemotherapeutic agents, such as cisplatin, doxorubicin, and histone deacetylase inhibitors, etc(37). In addition to AKT3, we in this present study also found that mTOR and its downstream target molecules p70S6K and eIF4EBP1 were all phosphorylated and activated by LINC00680 overexpression, whereas knockdown LINC00680 significantly inhibited their phosphorylation, suggesting that mTOR mediated p70S6K and eIF4EBP1 activation might play an integral role for transduction of LINC00680/miR-568/AKT3 signaling and the corresponding modulation of tumor stemness and chemosensitivity in HCC.
{kind=link}
{kind=link}